
PhD protein design @ColumbiaMed | prev bio @Lux_Capital, ML for Cas proteins @arcinstitute @UCBerkeley🐻 | ❤️🔥a
Joined March 2021
- Tweets 210
- Following 650
- Followers 406
- Likes 1,448
7 Photos and videos

Matthew Nemeth retweeted

Jun 12
The analogy with Moore’s law is weak because the curve tracks progress in a measurement tool weakly coupled, and sometimes irrelevant, to most end goals in biotech. Even in the subset of cases where it’s directly used, such as prenatal screening, still don’t see broad adoption of the best sequencing technology (which you would expect immediately) because the rate-limiting step is elsewhere: generally regulation, reimbursement. The curve is often used to paint a misleading picture of where the bottlenecks are, and can be used to sell or build products around constraints that are not actually rate-limiting.
Jun 12

Hard to think of another chart in biology more misunderstood and misappropriated

5
5
52
10,189
Matthew Nemeth retweeted

May 27
I have long written and spoken about the many ways US immigration policy harms international students and scholars. This has been true for as long as I can remember, including when I first came to the US on a single-entry student visa more than 20 years ago under a process formerly known as muslim registry program.
But the current administration has gone much further, through arbitrary policy changes, travel bans, and broad visa processing pauses that leave folks unable to work, travel, or train.
These policies affect a minority of scientists, and in the current state of the world they can be easy to overlook. But we should not let that happen.
I wrote about the quiet loss of Iranian scientific talent in US labs for @TheScientistLLC:
the-scientist.com/the-quiet-…
342
111
496
278,883
Matthew Nemeth retweeted
May 28
Biology is the next agentic frontier after coding. Anthropic is aggressively improving their models on routine data analysis with careful attention to nuances of different assay types. Opus 4.8 is noticeably better at single cell / spatial analysis. We have already rolled it out to customers across pharma and academia. Cool to see our benchmarks on the system card.


Introducing Claude Opus 4.8: it builds on Opus 4.7 with sharper judgment, more honesty about its own progress, and the ability to work independently for longer than its predecessors.
Available today at the same price.
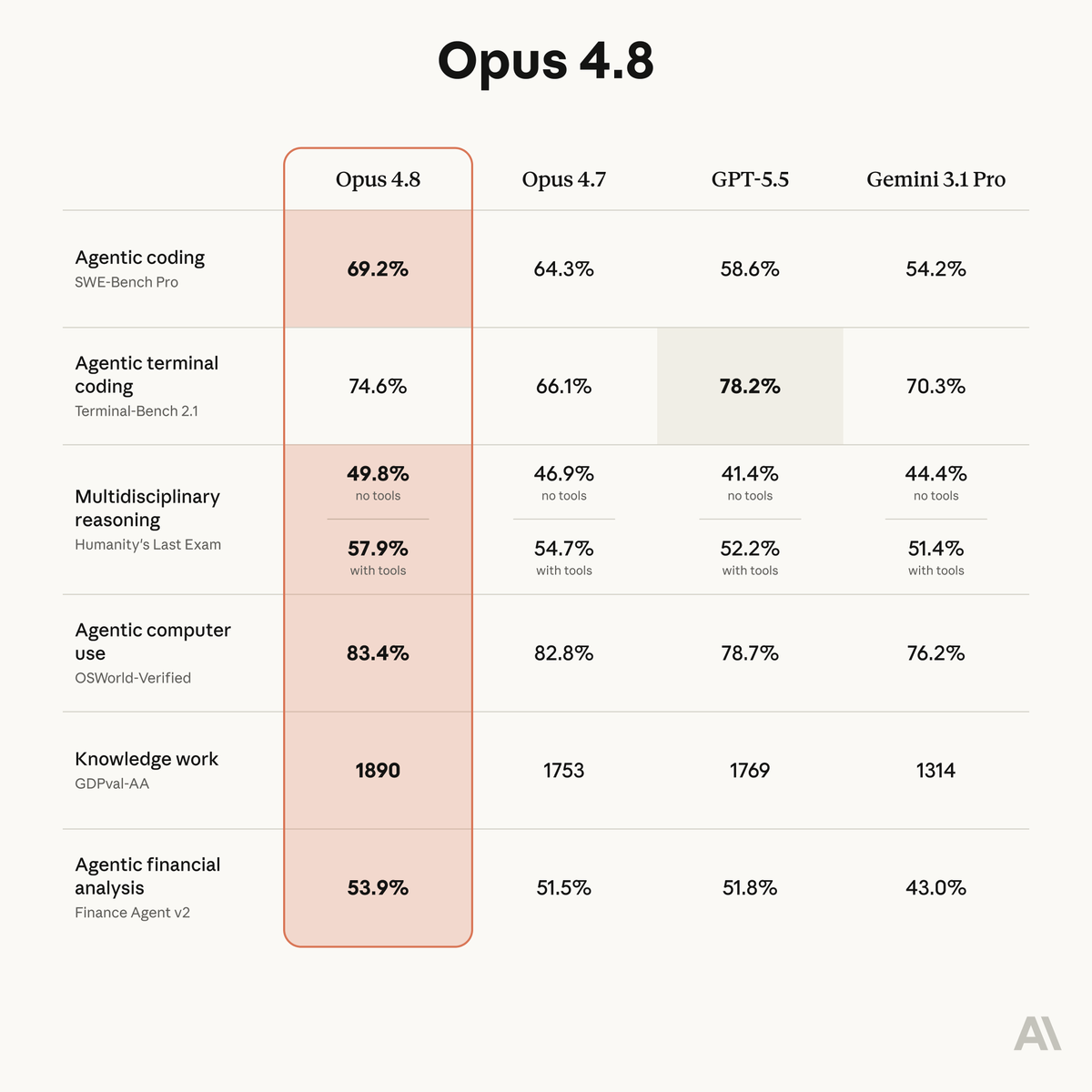
ALT Benchmark table showing how Claude Opus 4.8 compares to its predecessor and to other models on tests of coding, agentic skills, reasoning, and practical knowledge work tasks.
8
41
284
21,889
Matthew Nemeth retweeted
May 27
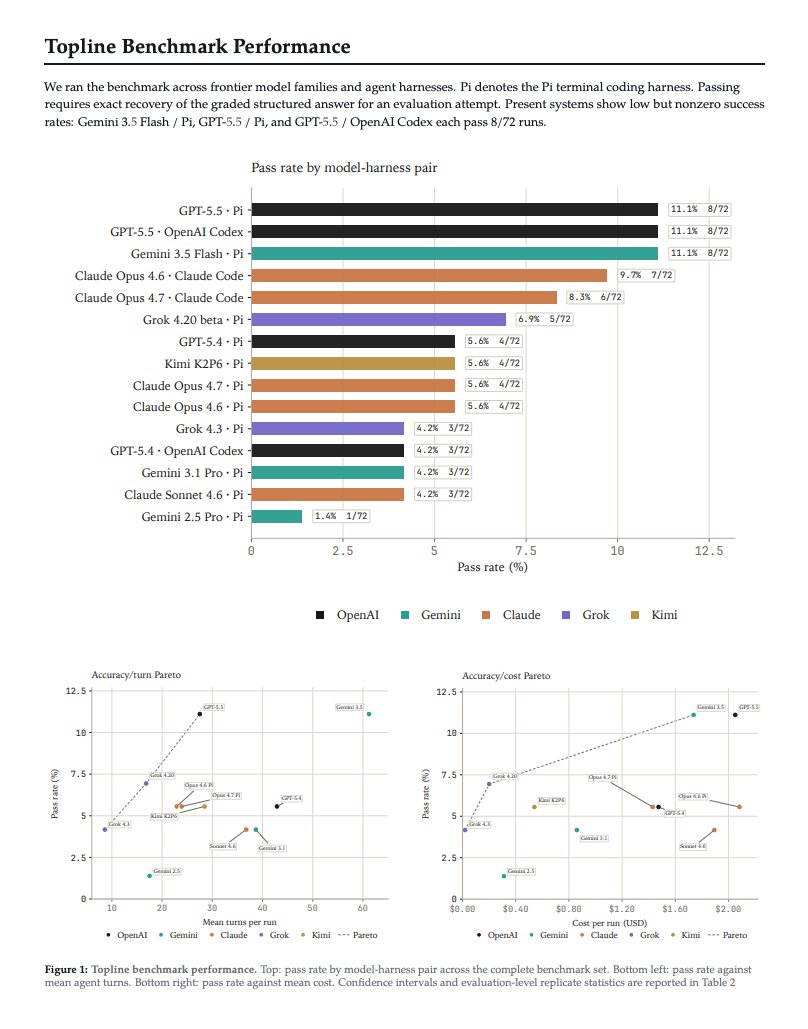
Introducing SpatialBench-Long, a benchmark for long-horizon spatial biology. Agents must recover biological claims from raw data and realistic experimental context without prescribed methods.
24 evals span primary tumors, organoids, xenograft models, lineage-tracing systems, and aging/intervention biology. The best agents score 11.1%.
1
17
67
8,485
Matthew Nemeth retweeted
May 27
Figuring out how to benchmark agents on realistic biology research has quickly become one of my favorite types of engineering work. You work with scientists to get to the core of some biological claim, precisely assembling raw data/prior literature/experimental context in a little 'world' for an agent to stumble around in - only for its behavior to challenge what you thought to be true and to force you to deeply introspect empirical human behavior. Doing this well gets considerably more difficult as we better approximate and climb long horizon scientific work: the type one might publish in the results section of a paper or build a drug program around. Been working on a project in this direction for the past few months and excited to drop tomorrow.
2
12
90
4,465
Matthew Nemeth retweeted

May 21
Thrilled to share I've started at @Stanford's Department of Pathology (@StanfordPath) in addition to @ArcInstitute. Looking forward to a shorter commute after 5 years at @BerkeleyBioE and embarking on daring new projects
We're recruiting multiple postdocs and technical staff👇
30
32
647
52,668
Matthew Nemeth retweeted

May 8
Introducing Genie 3, a generative protein model that substantially advances the state-of-the-art for binder design, increasing in silico success rates by up to 20x on hard multimeric targets. It also debuts a form of inference-time scaling unobserved in other design models. 🧵1/8
8
110
435
74,511

Genie3 is out! SoTA binder design, super fast inference & clean codebase.
I bet it’s a great base model for applications and further reward guided tuning/sampling studies.
May 8

Introducing Genie 3, a generative protein model that substantially advances the state-of-the-art for binder design, increasing in silico success rates by up to 20x on hard multimeric targets. It also debuts a form of inference-time scaling unobserved in other design models. 🧵1/8
1
7
75
4,907
Matthew Nemeth retweeted

Apr 21
Does AlphaFold’s latent space encode only the native state or something like a distribution over conformations? We begin to answer this question with ConforNets, a mechanism for producing diverse states, or very specific ones, via inference-time adaption of OF3p’s latent space👇

We introduce ConforNets, a mechanism for conformational control in AlphaFold3 models
- SoTA at producing diverse conformations on every multistate benchmark (N=104)
- Novel capability: transfer state from one protein to another
Outperforms BioEmu, ConforMix and AFsample3
🧵1/8
2
42
213
24,045
We introduce ConforNets, a mechanism for conformational control in AlphaFold3 models
- SoTA at producing diverse conformations on every multistate benchmark (N=104)
- Novel capability: transfer state from one protein to another
Outperforms BioEmu, ConforMix and AFsample3
🧵1/8
10
72
284
51,465
Matthew Nemeth retweeted
Mar 13
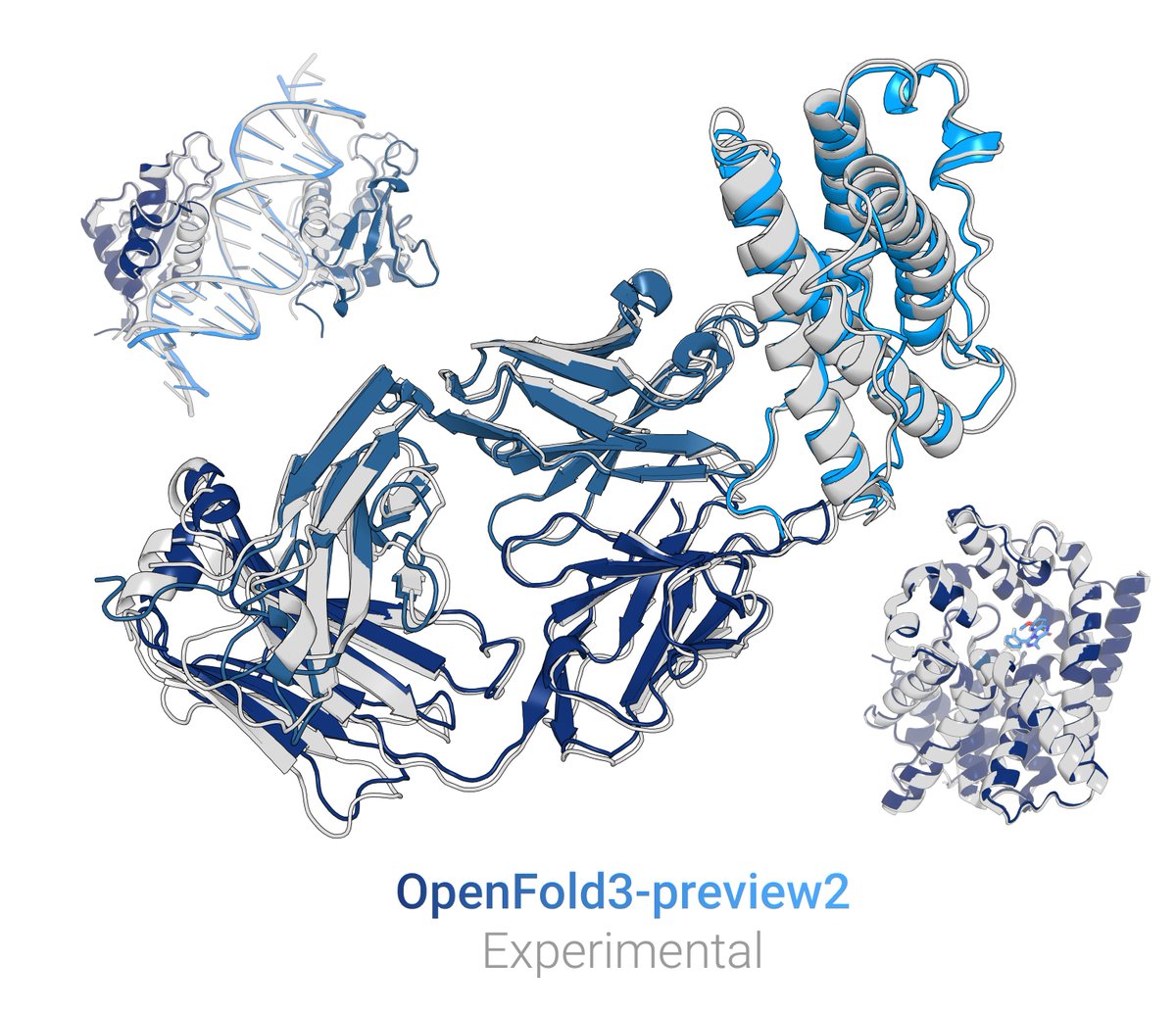
New OpenFold3 preview out! (OF3p2)
It closes the gap to AlphaFold3 for most modalities.
Most critically, we're releasing everything, including training sets & configs, making OF3p2 the only current AF3-based model that is functionally trainable & reproducible from scratch🧵1/9
8
178
675
55,771
Matthew Nemeth retweeted

Mar 10
First in Human! When @rhomsany and I first started Octant, this was the dream. A platform that makes molecules that few others can go after… to get the chance to tackle severe diseases with poor to no standard of care. It's been a long journey but so incredibly proud of the team and thankful to the volunteers who make this attempt possible. We got to celebrate with these new sunhats! octant.bio/news/octant-annou…

36
20
244
23,321

Matthew Nemeth retweeted

Feb 20
Must read paper 📝
Feb 19

We mapped gene interactions across different environmental conditions (GxGxE) at scale for the first time in human cells. These maps lead to the realization that many genes function in a context dependent manner which provides insight into how humans have relatively few genes but many cell types. Congratulations Ben!
Paper:
cell.com/molecular-cell/full…
1
1
449
Matthew Nemeth retweeted

Feb 19
Excited to see this out! Grateful to Patrick for his mentorship and support throughout the arc of this story.
We look forward to seeing what the community will engineer with MULTI-evolve!
Feb 19

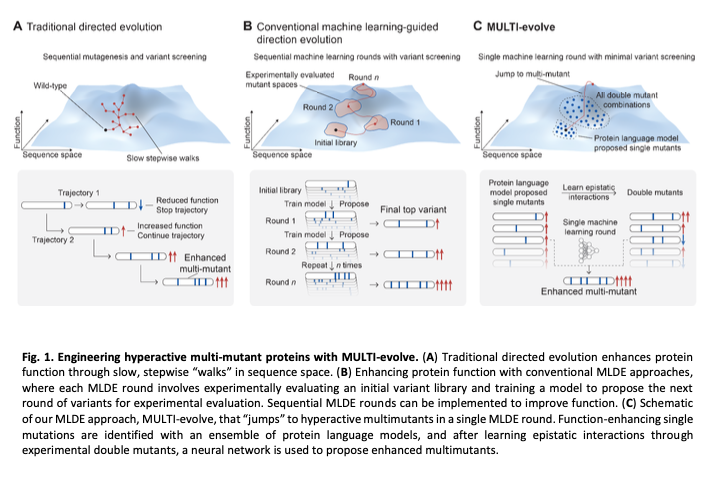
Delighted to share new @arcinstitute work from our group on AI-accelerated lab-in-the-loop, in @ScienceMagazine today
One of the most remarkable things about biology is that it's digital. DNA, RNA, proteins: these are all sequences, and their function is directly encoded in their sequence of letters. But a protein of length N has 20^N possible variants and the vast majority are non-functional. Evolution spent billions of years finding the functional needles in this haystack through random exploration and natural selection. For modern biomedicine, we need to solve this in days to weeks.

1
1
19
2,320
Matthew Nemeth retweeted
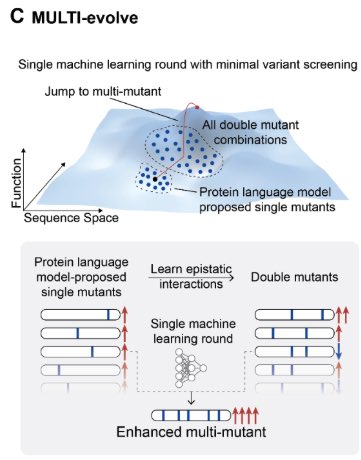
Feb 19
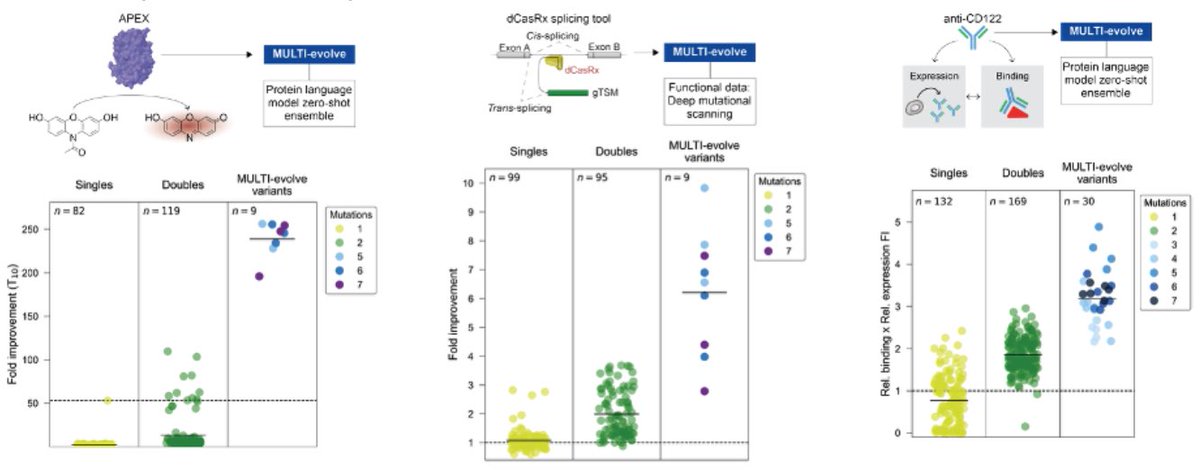
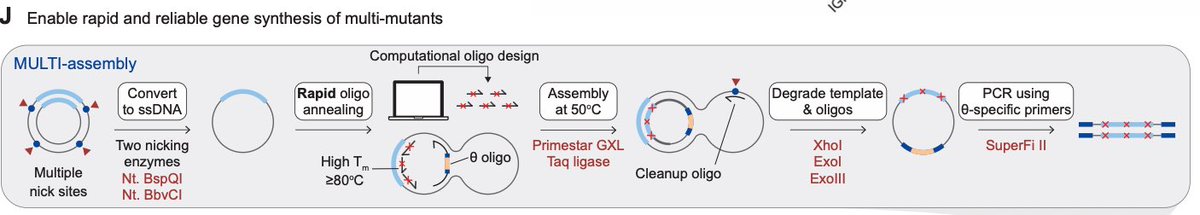
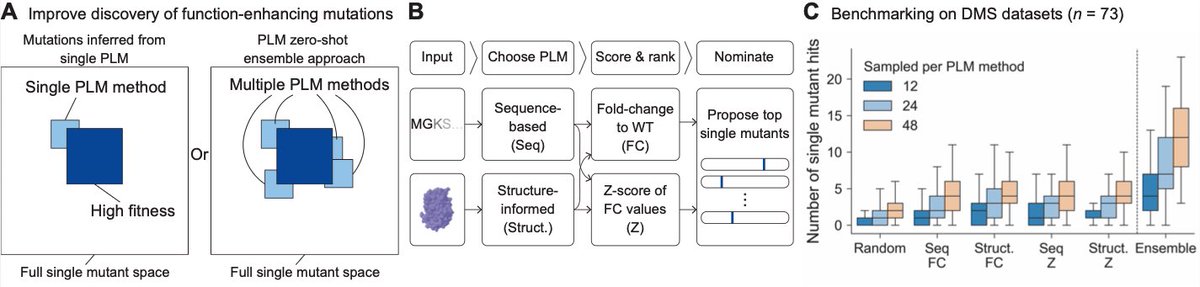
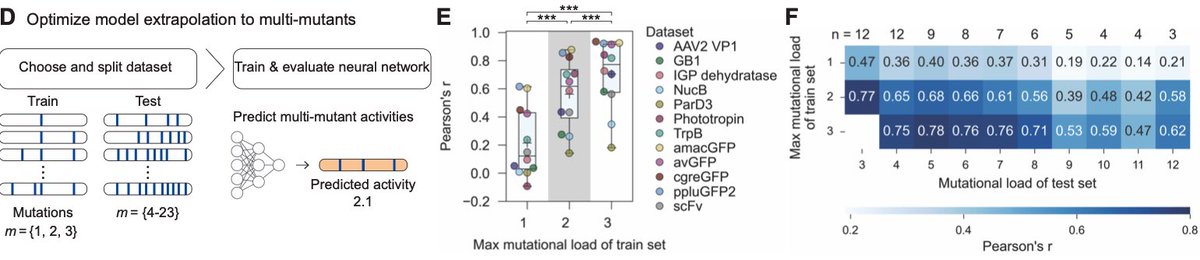
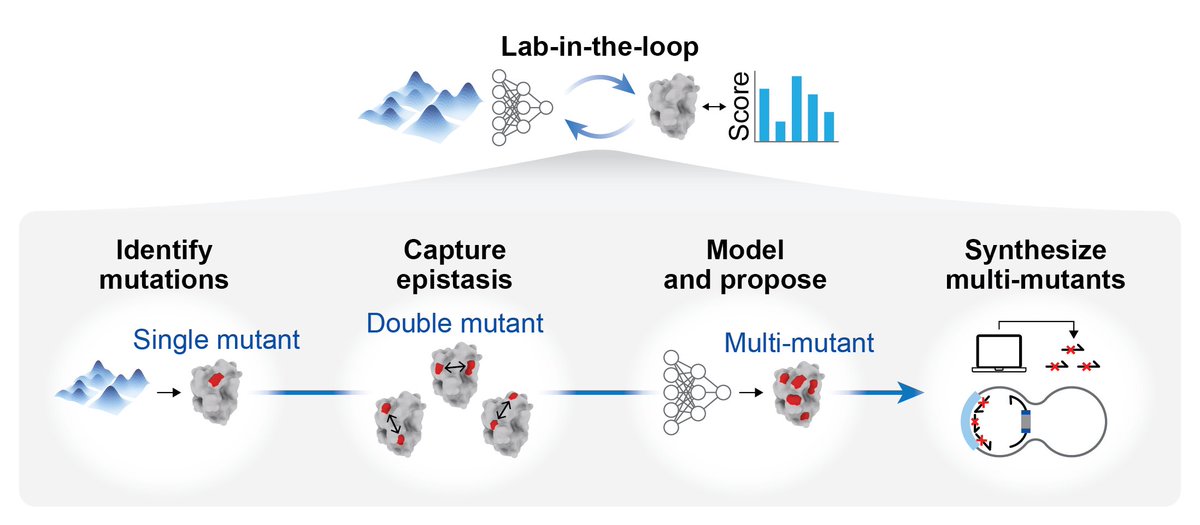
MULTI-evolve is one of our first answers. It's a full-stack, AI-lab-in-the-loop framework that "jumps" directly to hyperactive multi-mutant proteins via ML-guided evolution. We combine an ensemble of protein language models pretrained on all proteins across evolution to discover beneficial mutations, then systematically measure pairwise combinations to learn the epistatic landscape (e.g. which mutations are synergistic vs. antagonistic), and extrapolate to predict powerful higher-order combinations of 5-7 mutations. We also built MULTI-assembly, a molecular biology method to physically construct these complex multi-mutants cheaply and quickly, regardless of protein length (previously a major bottleneck).
1
2
60
5,679
Matthew Nemeth retweeted
Feb 19
The process of scientific research is fundamentally a search problem, and we basically do guess and check. We've trained predictive models of biology, like our Evo series of DNA language models, to learn the evolutionary constraints on biological sequences. Such models learn a fitness landscape of what evolution has explored. But the fitness landscape is not the same as the function you actually care about: whether an enzyme catalyzes faster, whether an antibody binds tighter, whether a CRISPR tool edits better. The core question is how do you connect the knowledge of these models to the functional search that has to happen in the physical lab?
2
4
85
7,079
Matthew Nemeth retweeted
Feb 19
Delighted to share new @arcinstitute work from our group on AI-accelerated lab-in-the-loop, in @ScienceMagazine today
One of the most remarkable things about biology is that it's digital. DNA, RNA, proteins: these are all sequences, and their function is directly encoded in their sequence of letters. But a protein of length N has 20^N possible variants and the vast majority are non-functional. Evolution spent billions of years finding the functional needles in this haystack through random exploration and natural selection. For modern biomedicine, we need to solve this in days to weeks.
18
135
697
91,936
Matthew Nemeth retweeted

Feb 19
Directed evolution revolutionized protein engineering, but still requires lots of time, iteration, and cost.
Today in @ScienceMagazine we share MULTI-evolve: our lab-in-the-loop approach with thoughtful integration of modern ML to make jumps across the fitness landscape.
5
32
196
10,963
Feb 19
Niko is so good at explaining…
Feb 19

Another interesting paper from @arcinstitute. This one combines protein language models with directed evolution to rapidly engineer proteins.
Here is how it works, step-by-step:
1. Select the protein you want to engineer. Give the protein sequence to four different protein language models which, together, "score" the likely fitness of each amino acid mutation. Get a ranked list of 50-100 protein mutations that these models *predict* might improve function. (This is all done on the computer.)
2. Take the top 15 predicted "hits" and then synthesize the pairwise combination for each of them. For example, if a mutation at amino acid #100 boosted activity, but so did mutations at positions #120, #135, and so on, then you'd make a protein with each of these "double" mutations. If you took the top 15 hits, then this is only 105 total proteins to synthesize.
3. Make and test all the double mutants in the wet-lab. Measure the activity of each double mutant. So, for example, if you wanted to engineer a protein to be "brighter," then you would put each double mutant in a microplate well and measure this directly. (This step basically captures epistatic relationships; it helps the models figure out which mutations are beneficial or damaging.) Feed the single double mutant activity data to a neural network, called MULTI-evolve. The model extrapolates these data to infer *additional* mutations that might be synergistic, like combinations of 5-7 amino acid swaps.
4. Take the top three predictions for proteins with 5-, 6-, or 7 amino acid mutations, based on the neural network. Synthesize these proteins using a new DNA assembly approach, also reported in this paper, called MULTI-assembly. (The gist is that you anneal together a bunch of short oligos, each carrying one of the mutations, in a tube to reconstruct each of the full genes. This yields correctly built sequences 40-70% of the time.)
5. Finally, express the proteins in cells, measure their activities, and benchmark them against the wild-type protein.
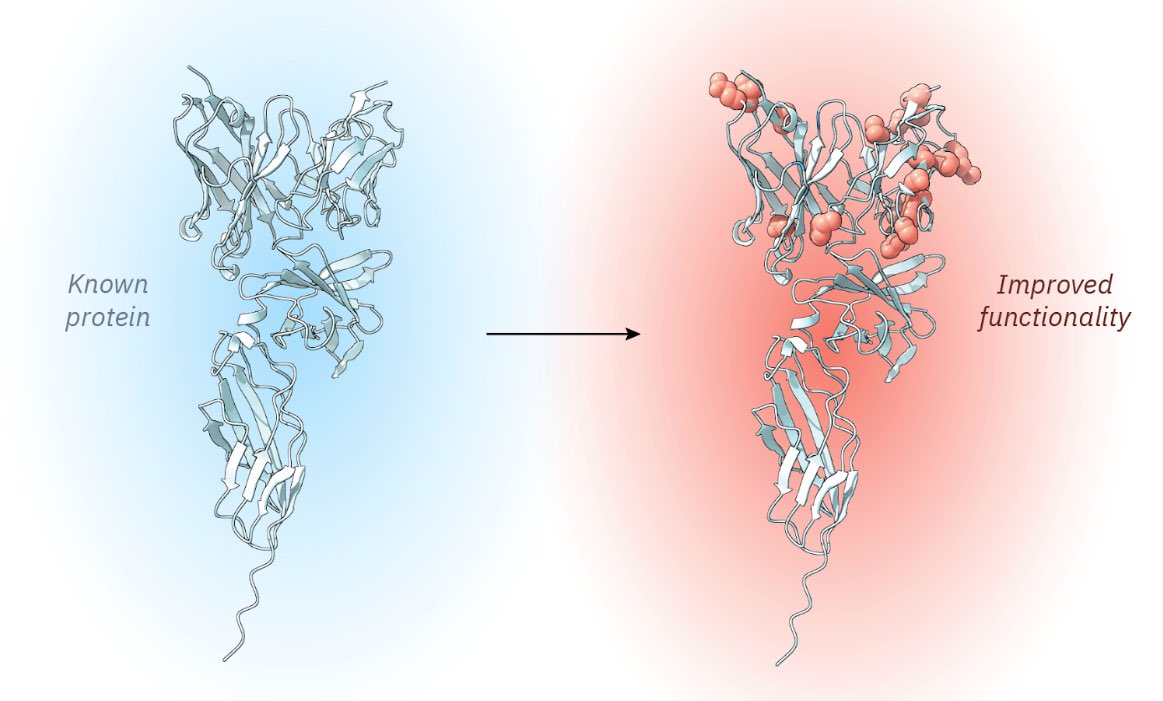
The researchers used this method for various proteins. For one protein, called dCasRx (a Crispr protein that targets RNA instead of DNA), they used it to create a variant with 9.8-fold better activity, and they validated this across three different human genes.
You can also optimize proteins for two different properties at the same time. The authors used their method, for example, to engineer an antibody targeting CD122 for both binding affinity AND its expression yield in cells.
TL;DR This is a new way to speed up directed evolution. Instead of using random mutations to search through a huge amount of "biological space" (remember that a single protein, with just 100 amino acids, has 20^100 possible combinations), these researchers use AI models to navigate this search space for proteins more quickly. Can we use the same approach to make entire gene circuits?

8
2,916