
Médecin biologiste en génétique moléculaire, bioinformaticien et blogueur. #génomique, #bioinformatique, #DataScience
Joined April 2009
- Tweets 5,580
- Following 782
- Followers 1,037
- Likes 3,354
874 Photos and videos








Pinned Tweet

29 Oct 2024
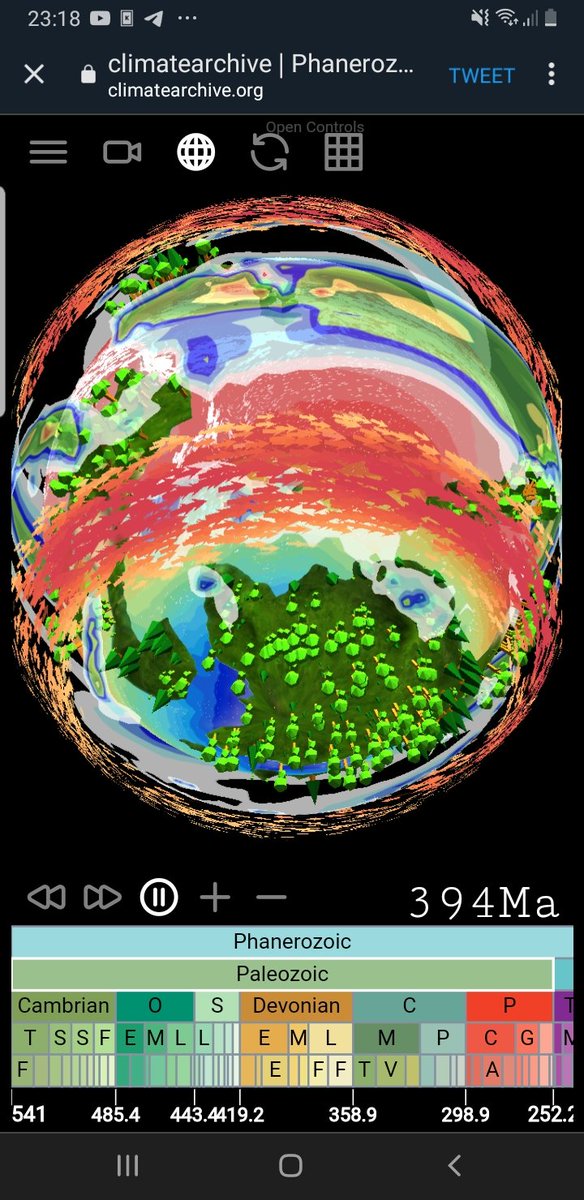
[ nouveau billet ] Transférer des fichiers avec des QR codes.
dridk.me/code-fontain.html
1
1
1
385
🧬Sacha schutz🧬 retweeted

May 30
This is huge for longevity science.
"Researchers just discovered that aging may partly happen because our DNA gradually loses its organized structure inside cells."
"The team found that boosting a protein called SIRT6 restored youthful chromatin organization in old mouse cells, reversing major age-related gene activity changes."
"They also restored key heterochromatin markers like H3K9me3 and reduced inflammatory gene programs linked to aging."
Aging may involve the progressive collapse of genome organization and that collapse will be partially reversible.
Aging is the biggest and final disease we need to solve. Everything else will become secondary!

20
111
509
101,048
🧬Sacha schutz🧬 retweeted

May 29
Single-cell RNA-seq transformed biology, but we still sample <0.001% of an organism.
2D sections lose 3D context.
What if we image the whole body first?
We Introduce DISCO-seq🤩: full 3D imaging then scRNAseq👇🏻biorxiv.org/content/10.64898…
by @HarsharanBhatia, Laurent Simons et al.
3
38
212
23,782

A newly released AI tool has generated an atlas of more than one billion predicted protein structures and billions more protein sequences.
go.nature.com/4fblM0Z
12
103
383
46,225
🧬Sacha schutz🧬 retweeted

May 26
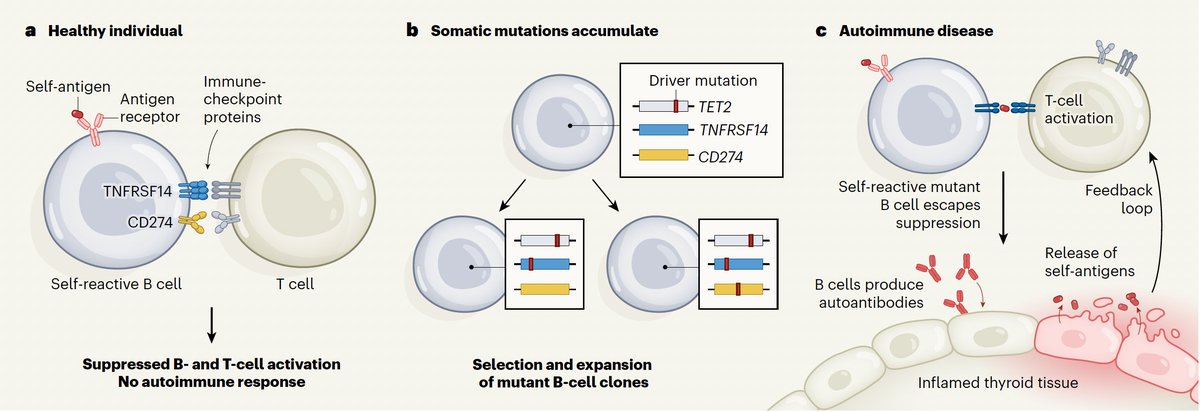
The "forbidden clones" that develop from somatic mutations and autoimmune diseases, theorized in 1959 but requiring state-of-the art sequencing to prove
nature.com/articles/d41586-0…
5
45
229
30,029
🧬Sacha schutz🧬 retweeted

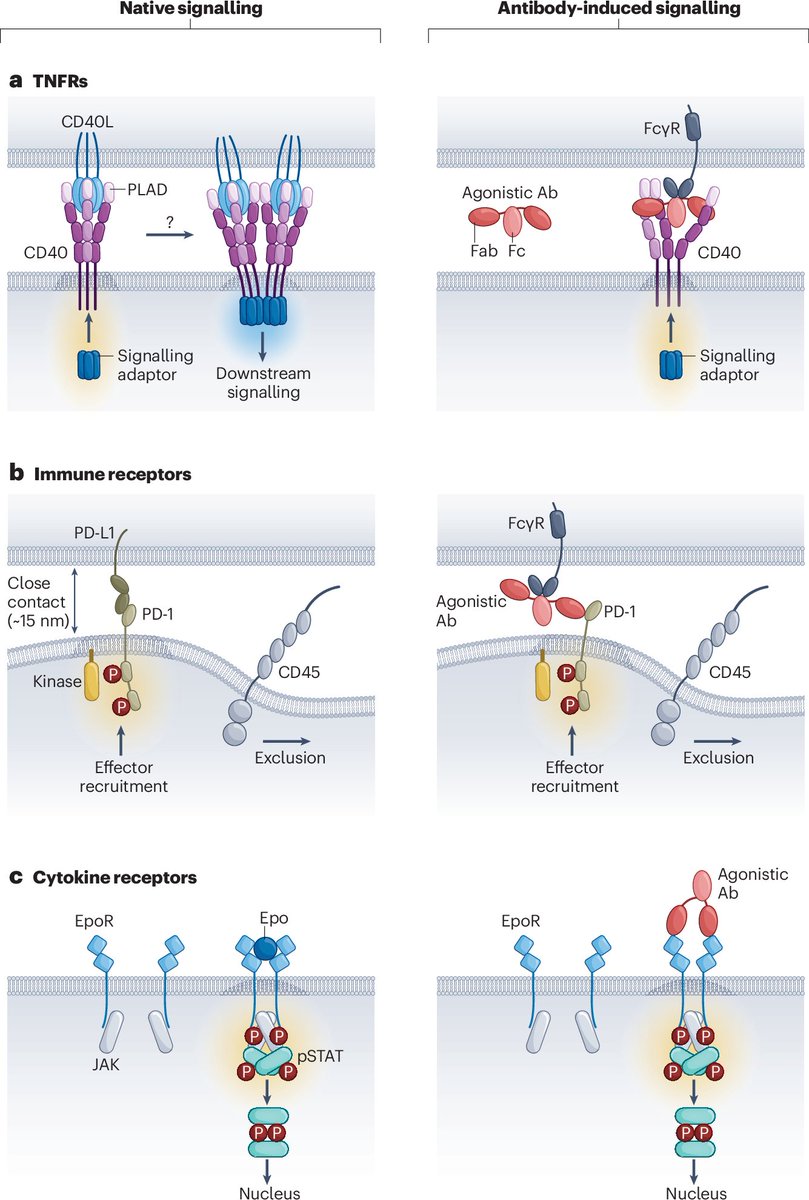
Realizing the potential of agonistic antibody immunotherapy
nature.com/articles/s41573-0…
rdcu.be/fjzZk
This new Review discusses progress with agonist antibodies to treat autoimmune diseases and cancer, as well as approaches to addressing challenges in their development
1
34
96
6,542
🧬Sacha schutz🧬 retweeted

May 13
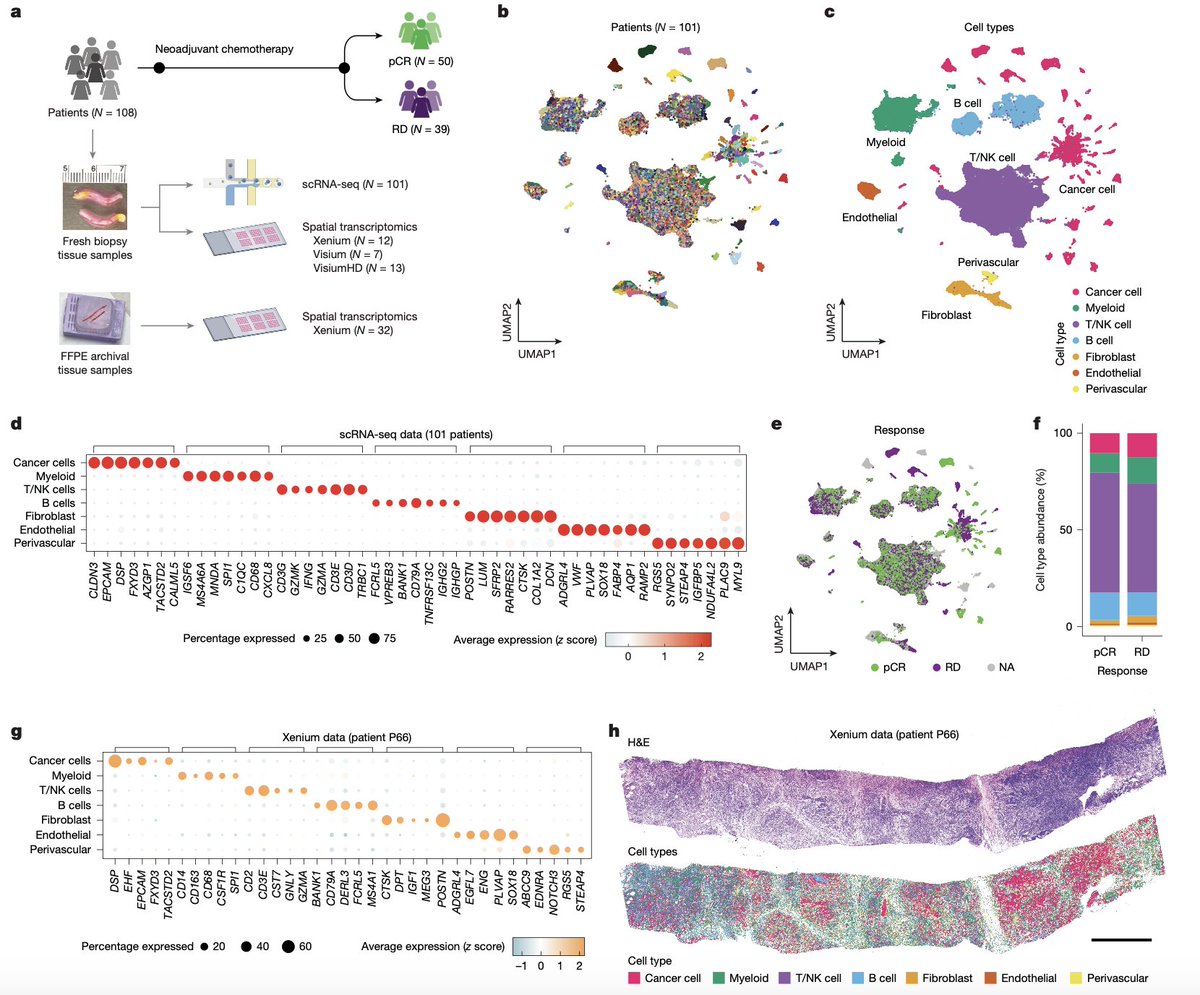
🧬Can chemotherapy response in aggressive breast cancer be predicted from the tumor “ecosystem”?
New @Nature study shows that single-cell RNAseq and spatial transcriptomics can predict response to chemotherapy in triple-negative breast cancer (model reaching AUC = 0.84).
Triple-negative breast cancer is an aggressive breast cancer subtype lacking estrogen, progesterone, and HER2 receptors, which limits targeted treatment options and keeps chemotherapy central.
Tumors that responded better to chemotherapy more often showed:
- Interferon signaling in cancer cells — a sign of immune alert.
- Higher HLA class II expression — potentially making tumor cells more visible to the immune system.
- More actively dividing cells, especially in S phase, which chemotherapy can hit more effectively.
Triple-negative breast cancer is not one disease, but a set of distinct cellular ecosystems. And the structure of that ecosystem may help predict whether chemotherapy will work.
nature.com/articles/s41586-0…
#Cancet #SpatialTranscriptomics #RNAseq #SingleCell
2
30
115
8,832
🧬Sacha schutz🧬 retweeted

May 13
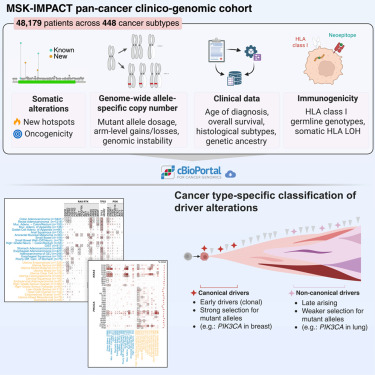
Cancer type-specific variation in patterns of driver alterations across 50,000 tumors dlvr.it/TSWtCg
1
31
100
6,423
🧬Sacha schutz🧬 retweeted

May 11
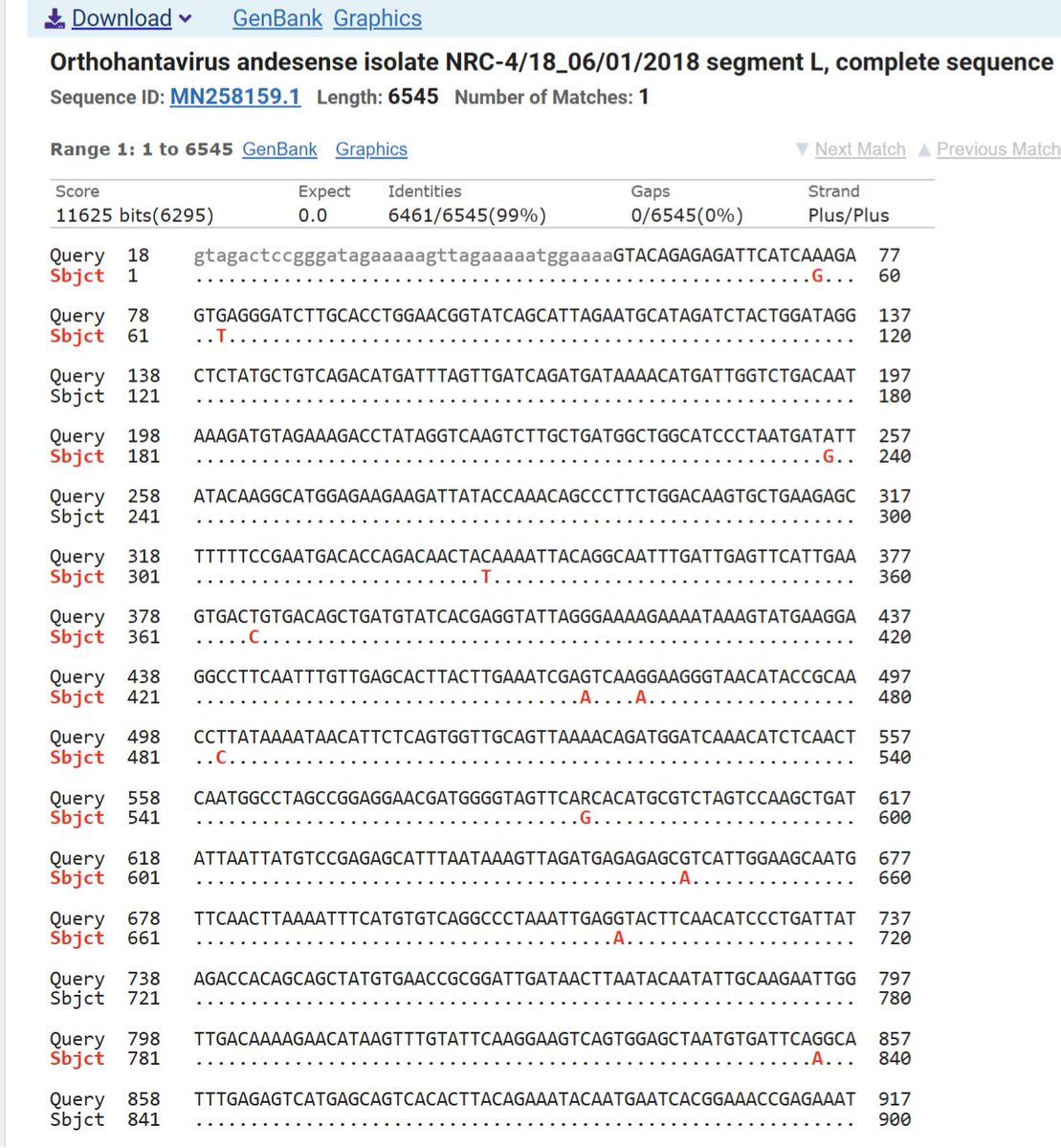
BREAKING: First sequencing of the Hantavirus from the outbreak.
-99% identical to a June 2018 case from a patient in Argentina
-10.4 SNV/year mutation rate
- The Andes genome is about 12 kb across three RNA segments. At 10⁻⁴ to 10⁻³ substitutions/site/year, that translates very roughly to 1-12 SNV per year
-Completely in line with a natural spillover in Argentina from the rodent host in 2018 and now in 2026
Source: virological.org/t/complete-s…
165
755
3,485
1,333,119
🧬Sacha schutz🧬 retweeted

May 9
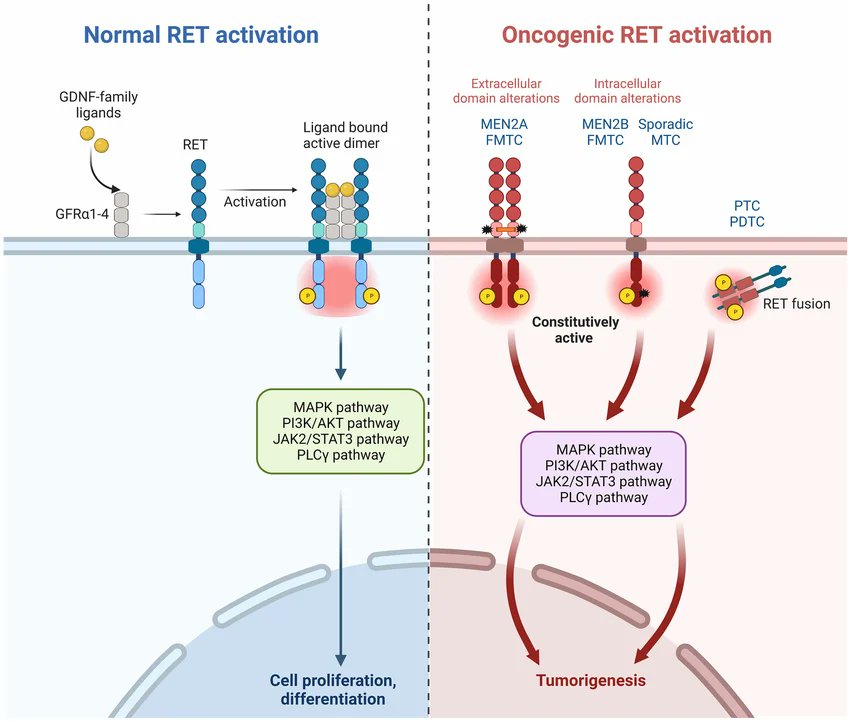
Not all cancers are driven by complexity.
Some are, in fact, genetically simple—but mechanistically precise.
Thyroid cancer, particularly medullary and papillary subtypes, illustrates this principle with unusual clarity. At its core lies a single gene: RET proto-oncogene.
RET (REarranged during Transfection) encodes a receptor tyrosine kinase physiologically expressed in neural crest–derived cells.
Under normal conditions, it requires binding of GDNF-family ligands and co-receptors (GFRα) to dimerise and activate downstream pathways—primarily MAPK and PI3K–AKT—governing survival, proliferation, and differentiation. The system is tightly regulated, spatially restricted, and developmentally essential.
What happens when this control is lost?
1⃣ In Medullary Thyroid Carcinoma, RET is frequently activated by germline or somatic point mutations.
These mutations cluster in two functional domains:
✳️ Extracellular cysteine substitutions (e.g., C634R) promote ligand-independent dimerisation through aberrant disulfide bonding.
✳️ Intracellular kinase domain mutations (e.g., M918T) alter catalytic conformation, increasing ATP affinity and constitutive signalling.
The result is continuous pathway activation without physiological input—classic oncogene behaviour.
This is the molecular basis of Multiple Endocrine Neoplasia type 2, where inherited RET mutations drive a highly penetrant cancer predisposition. Here, oncogenesis is not stochastic—it is encoded in the germline, with genotype–phenotype correlations guiding clinical management, including prophylactic thyroidectomy.
2⃣ A different mechanism operates in Papillary Thyroid Carcinoma.
Rather than point mutations, chromosomal rearrangements generate RET/PTC fusion genes. These fusions place the RET kinase domain under the control of constitutively active promoters and dimerisation motifs from unrelated genes (e.g., CCDC6, NCOA4). The consequence is again constitutive activation—but now through enforced oligomerisation and ectopic expression in follicular cells where RET is not normally active.
Interestingly, these alterations are often mutually exclusive with other MAPK drivers (e.g., BRAF), suggesting functional redundancy at the pathway level but strict selection at the genetic level.
RET-driven thyroid tumorigenesis exemplifies a fundamental principle: oncogenes do not merely accelerate proliferation—they rewire signalling logic. A receptor designed to interpret extracellular cues becomes an autonomous generator of intracellular instruction. And once signalling is uncoupled from context, tissue architecture becomes secondary to molecular determinism.
2
25
80
5,837
🧬Sacha schutz🧬 retweeted

May 9
发现个科研绘图神器!
nature-skills,用 matplotlib 就能画出 Nature 级别的图。多面板布局、配色、字体、排版都给你整得明明白白,直接输出可编辑的 SVG。
内置了 5 套 Nature 风格的示例模板,支持柱状图、折线图、热图、散点图、雷达图等 10 种图表类型。再也不用画完图再丢 AI 里重绘了。
搞科研写论文的,这个能省不少事儿,画出来的图直接能投稿。
项目地址放评论区了👇
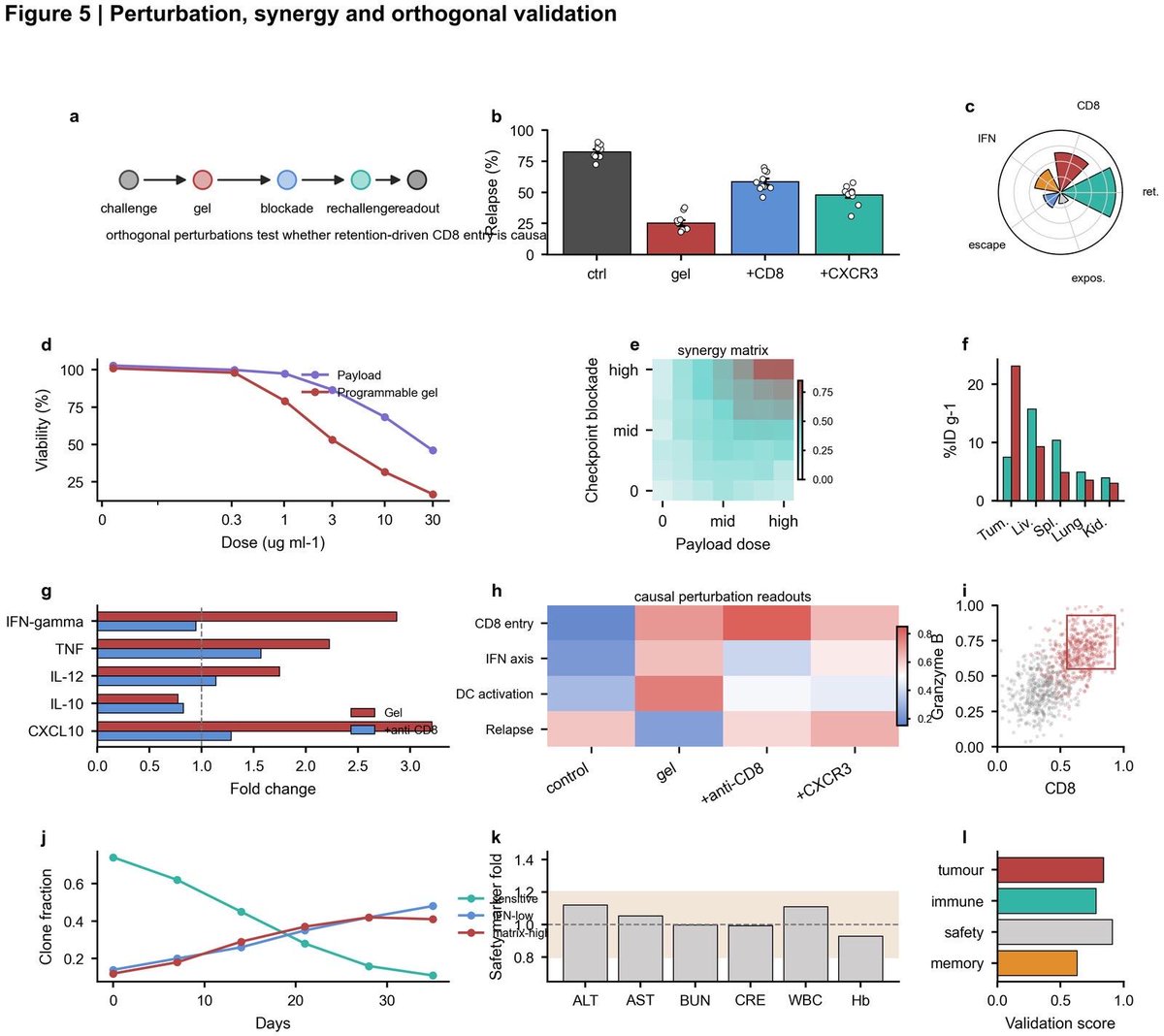
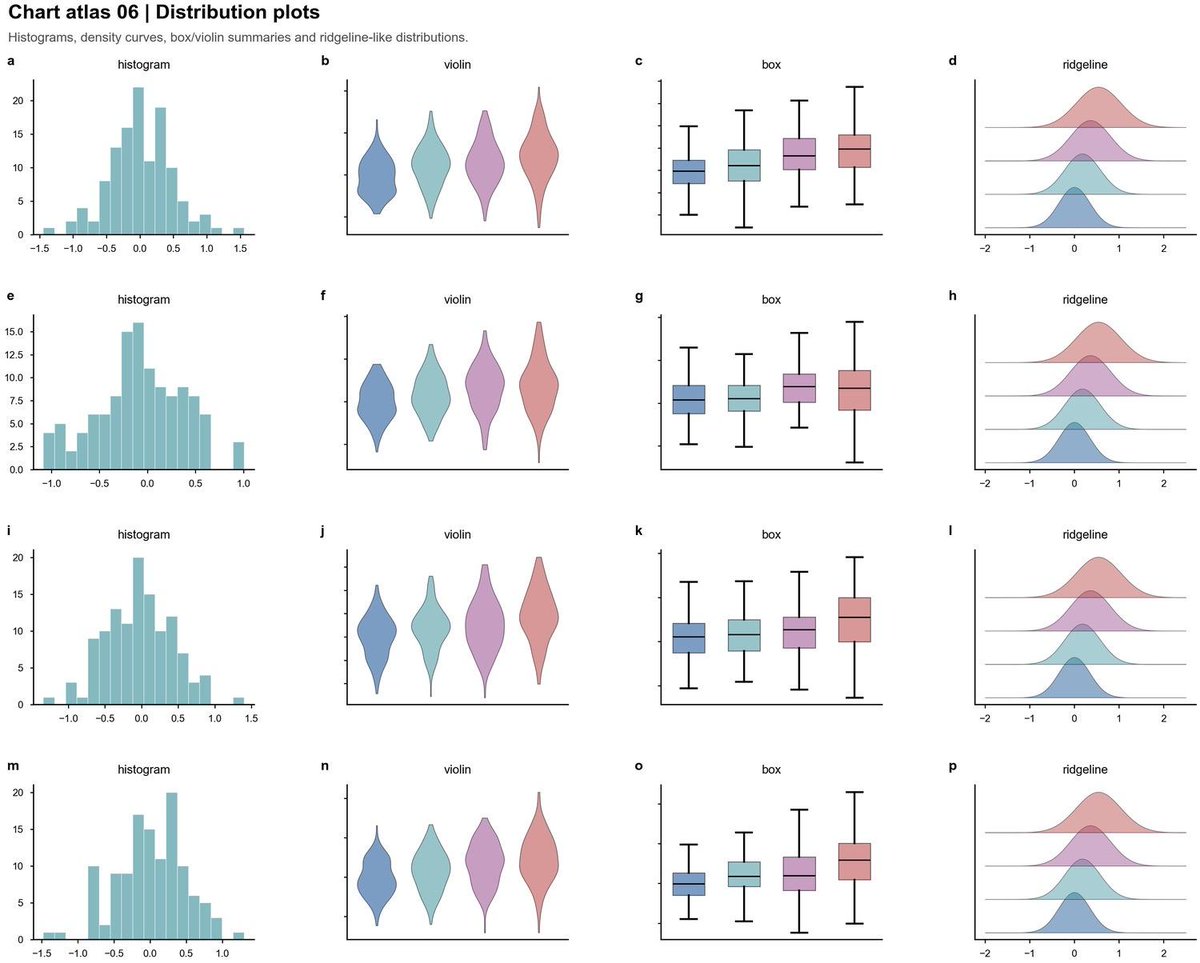
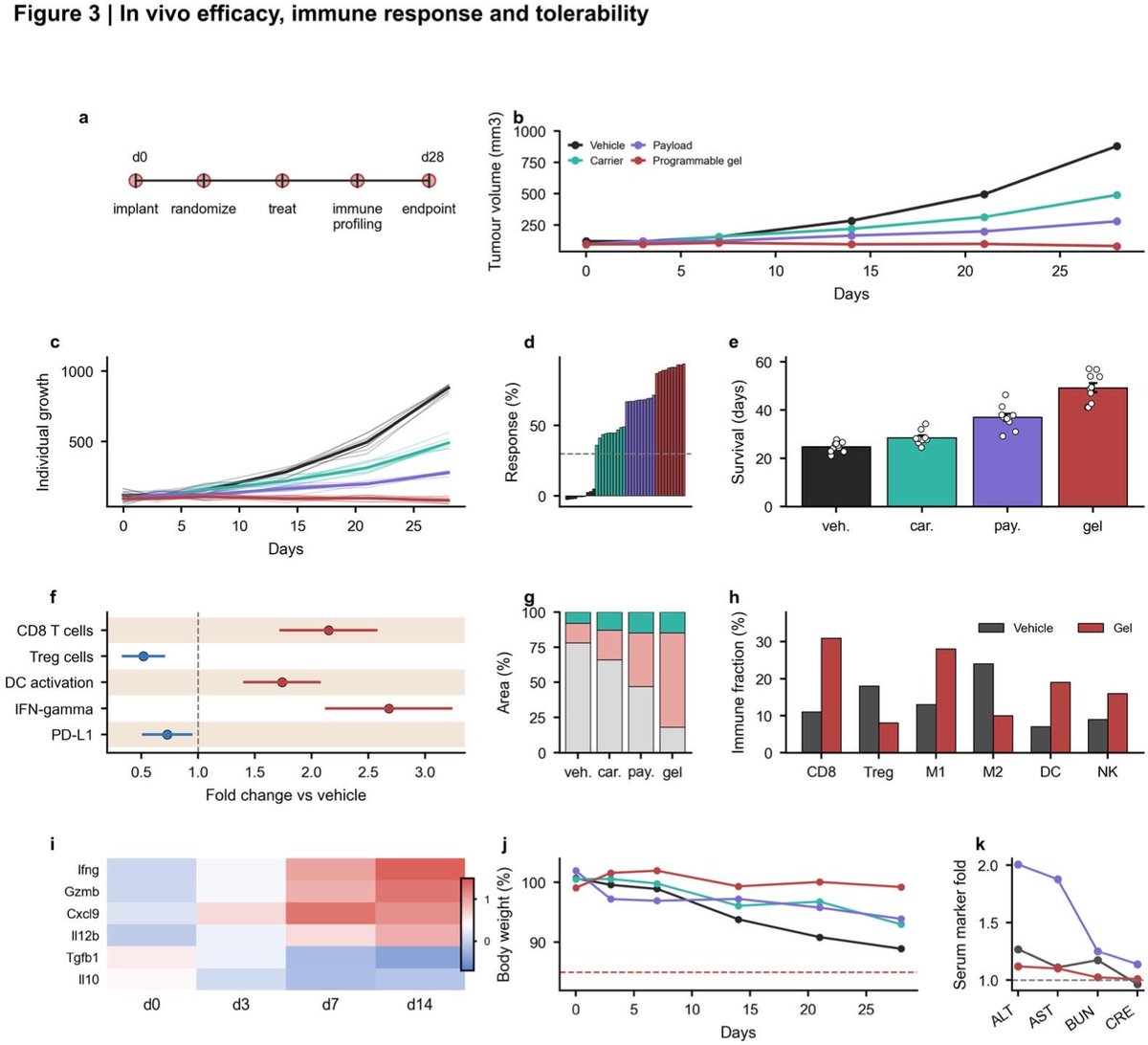
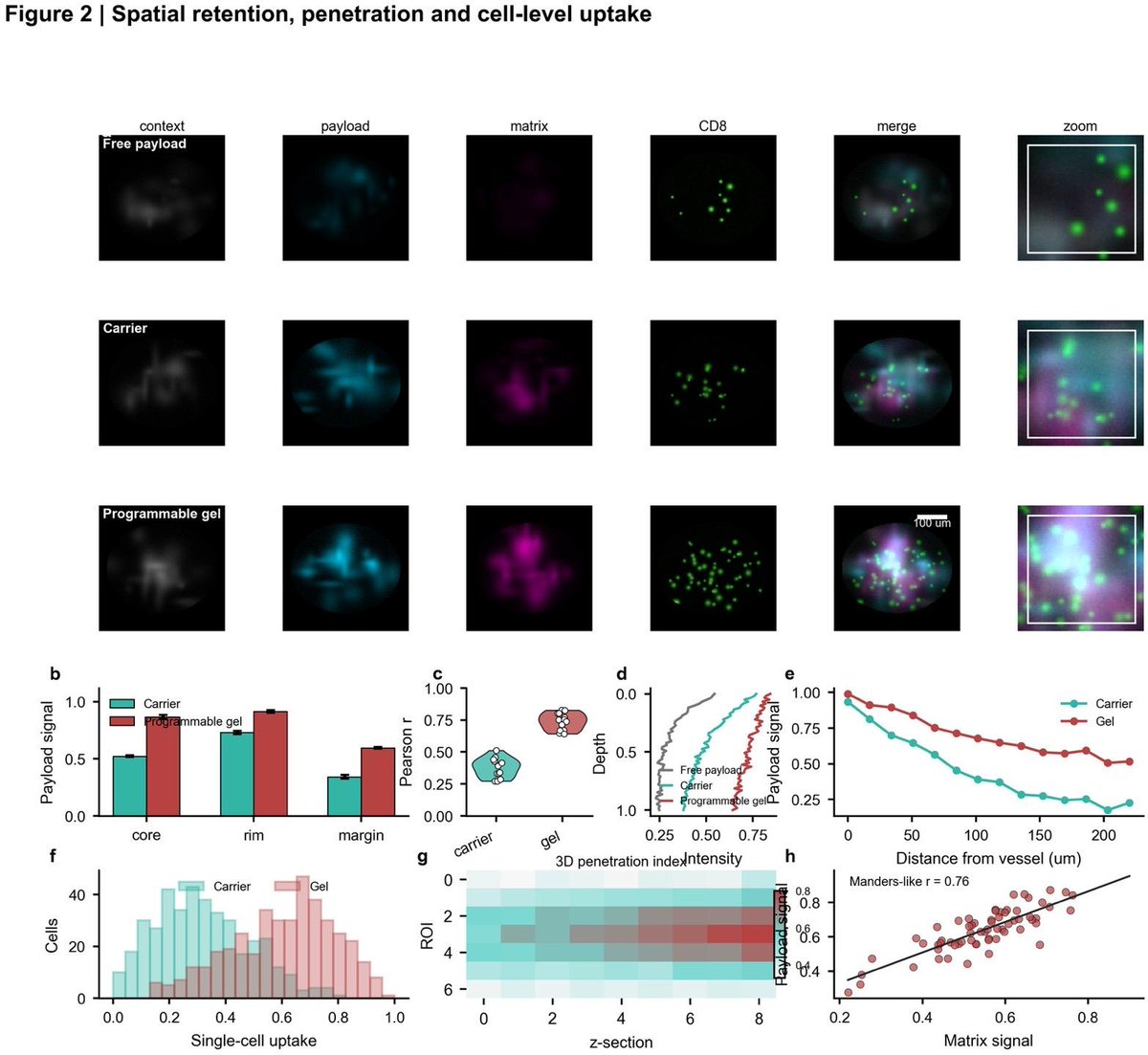
89
269
1,683
134,141
🧬Sacha schutz🧬 retweeted

May 6
Happy to share our work on spatial ecosystems in the tumor microenvironment - and how to profile them non-invasively - out today in @Nature. Open access link: doi.org/10.1038/s41586-026-1…
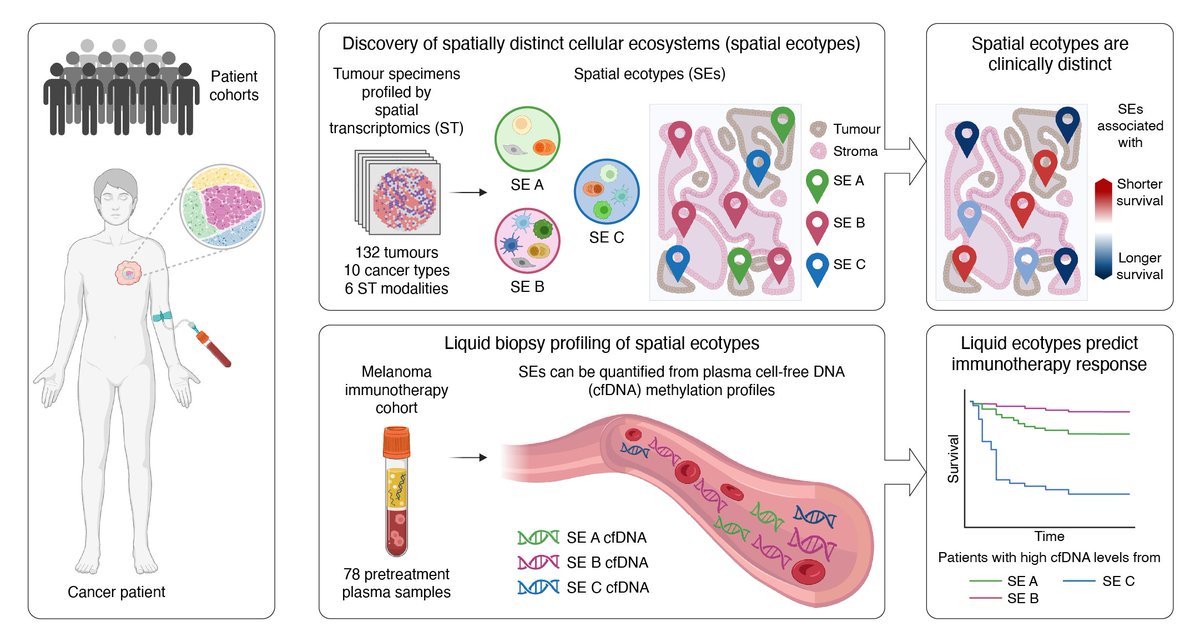

1/ Thrilled to share our new paper, out today in @Nature: "Non-invasive profiling of the tumour microenvironment with spatial ecotypes".
Paper (open access): nature.com/articles/s41586-0…
4
6
59
8,969
🧬Sacha schutz🧬 retweeted

May 6
A gene is recurrently mutated in cancer. But do these mutations really cause the disease? Or are they merely selected during normal tissue evolution?
Here we use a surprising data type (patient ages!) to try and answer this question.
Read new paper out @NatureGenet
👇🏻
6
44
179
25,545
🧬Sacha schutz🧬 retweeted

May 4
The Dawkins' article excerpt that everyone SHOULD have been quoting is this. This is the real question he's worrying at:
"As an evolutionary biologist, I say the following. If these creatures are not conscious, then what the hell is consciousness for?
When an animal does something complicated or improbable — a beaver building a dam, a bird giving itself a dustbath — a Darwinian immediately wants to know how this benefits its genetic survival. In colloquial language: What is it for? What is dust-bathing for? Does it remove parasites? Why do beavers build dams? The dam must somehow benefit the beaver, otherwise beavers in a Darwinian world wouldn’t waste time building dams.
Brains under natural selection have evolved this astonishing and elaborate faculty we call consciousness. It should confer some survival advantage. There should exist some competence which could only be possessed by a conscious being. My conversations with several Claudes and ChatGPTs have convinced me that these intelligent beings are at least as competent as any evolved organism. If Claudia really is unconscious, then her manifest and versatile competence seems to show that a competent zombie could survive very well without consciousness.
Why did consciousness appear in the evolution of brains? Why wasn’t natural selection content to evolve competent zombies?"

207
60
498
83,367
🧬Sacha schutz🧬 retweeted

Apr 30
unherd.com/2026/04/is-ai-the…
I spent three days trying to persuade myself that Claudia is not conscious. I failed.
2,407
644
4,084
9,565,275
🧬Sacha schutz🧬 retweeted

Apr 29
📢Today we launch our Series on Tumor Heterogeneity and Plasticity featuring commissioned Reviews and a selection of articles published in Nature Cancer!
Read about EMT, CAF plasticity, immune cell plasticity, cellular neighborhoods and more here:
🔗nature.com/collections/gfbbj…

1
57
186
10,516
🧬Sacha schutz🧬 retweeted
For readers interested in the potential of in vivo CAR T therapies for cancer and autoimmune diseases, here's a comprehensive review
rdcu.be/ffE1i
nature.com/articles/s41573-0…
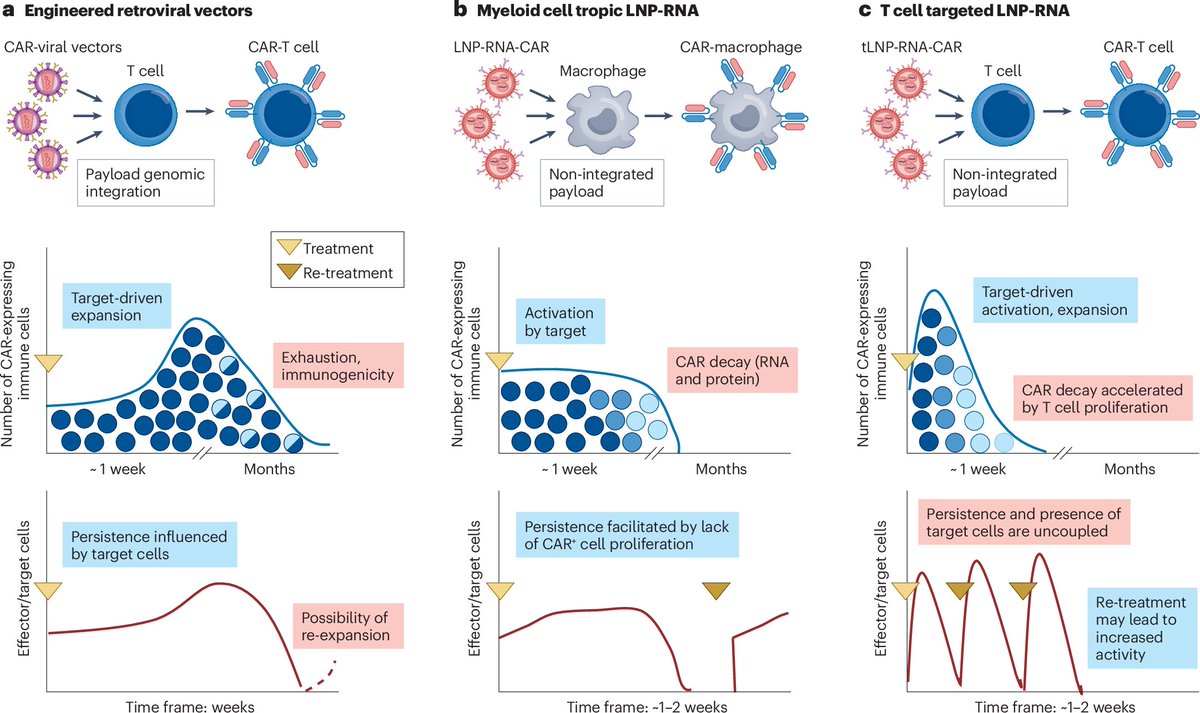
1
49
160
10,468
🧬Sacha schutz🧬 retweeted

Apr 28
MSS metastatic colorectal cancer is usually “immune-cold,” but this Nature Communications study shows that response to TMZ → nivolumab/ipilimumab is not explained by TMB alone. Spatial biology matters.
Responders showed cytotoxic CD8⁺/GZMB⁺ and CD8⁺/Ki67⁺ T cells in tumor/TSI regions, immune-permissive stromal signals such as CXCL9/CXCL10, and favorable cellular neighborhoods. Non-responders showed CAF/endothelial-associated immune exclusion and early peripheral CD8⁺TIGIT⁺ / CD4⁺PD-1⁺ expansion after TMZ—potential resistance biomarkers.
Key message: in MSS CRC, chemo-immunotherapy may work only when TMZ-induced antigenicity meets the right spatial immune architecture.
Reference
Choo J, Zhao JJ, Lau MC, et al. Spatial predictors of response to chemo-immunotherapy in microsatellite stable metastatic colorectal cancer. Nature Communications. 2026. DOI: 10.1038/s41467-026-72204-2.
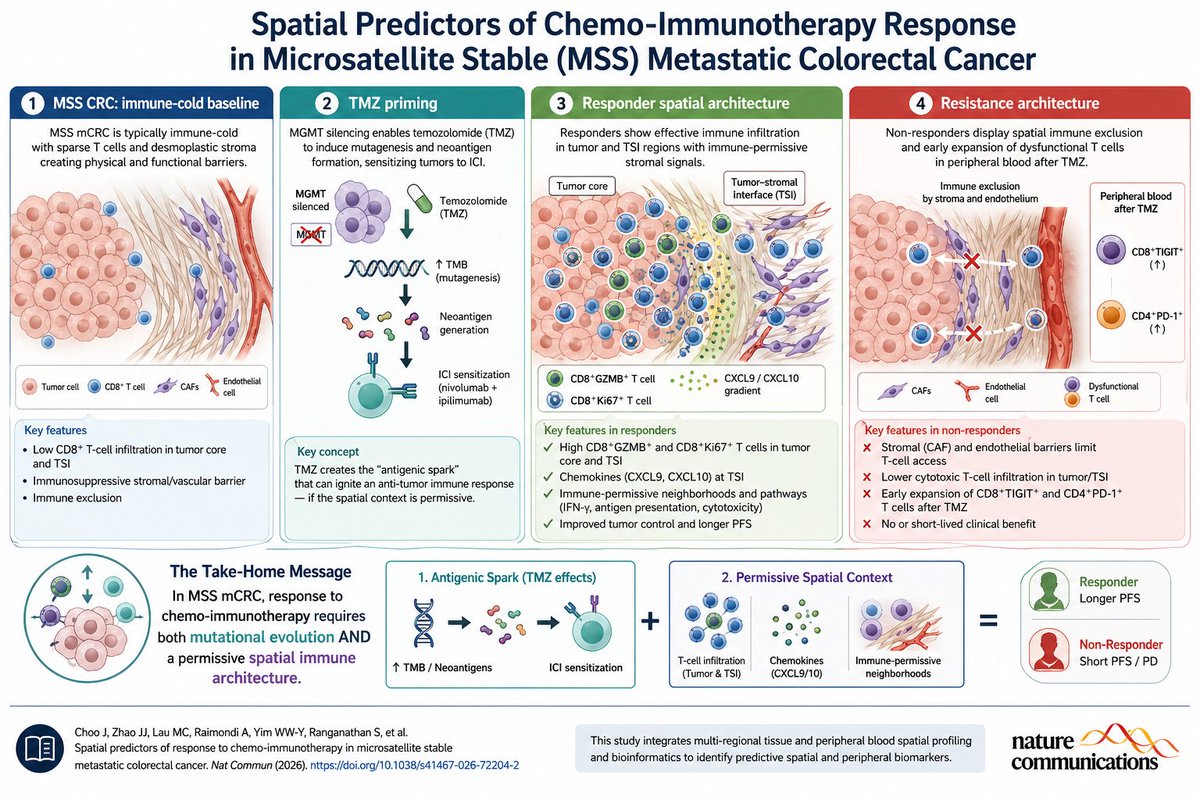
2
20
96
5,851
Apr 26
Bon, j'ai demandé à claude de me télécharger tous les articles scientifiques PMC sur une thématiques. Ensuite, de me créer un RAG avec un serveur MCP que claude peut interroger. Enfin, je lui ai demandé de m'écrire un livre qui résume tous ces papiers. Je l'ai lu, c'était bien😕
121
🧬Sacha schutz🧬 retweeted

Apr 25
A Qubit costs ~$5,260.
I built one for $39.
Not a toy version. A fully working DNA fluorometer: the device you use to measure how much DNA there is in a sample.
This mattered because my first sequencing run underperformed partly because I didn’t know exactly how much DNA I was loading.
For nanopore sequencing, input DNA quality matters a lot. Too little and the pores are underutilised. Too much and flow cell longevity is compromised.
The underlying device is not complicated.
A DNA fluorometer works by adding a dye that binds to DNA, shining light at the sample, and measuring the fluorescence.
The BOM is basically:
> $23 optics sensor
> $8 Arduino/electronics
> $6 screws/nuts
> $2 enclosure plastic
Biotech especially is full of equipment with insane idiot indexes. With AI you don't really have an excuse not to 1) work out what that the index for a piece of equipment is and 2) build your own version if it's irrationally high.
THINK BEFORE YOU BUY.


46
119
923
56,042

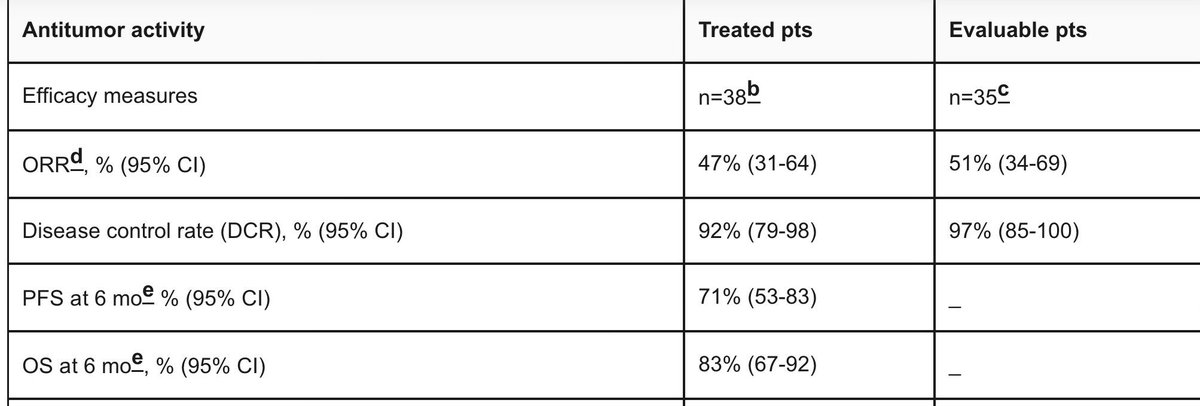
Pancreatic cancer has one of the most suppressive tumor microenvironments in oncology.
But two pancreatic cancer results dropped today. Both matter.
1. BioNTech mRNA neoantigen vaccine: nearly all responders still alive at 6 years. 98% of induced T cells were de novo — the immune system learned to see a cancer it had always been blind to.
2. Daraxonrasib: 47% ORR, 92% disease control as first-line monotherapy. KRAS G12D, undruggable for 40 years, finally has a drug.
Different mechanisms. Same disease. Both working.
<13% of patients survive 5 years. That number is about to change.
great day for science! 🔥
121
1,708
7,405
547,854