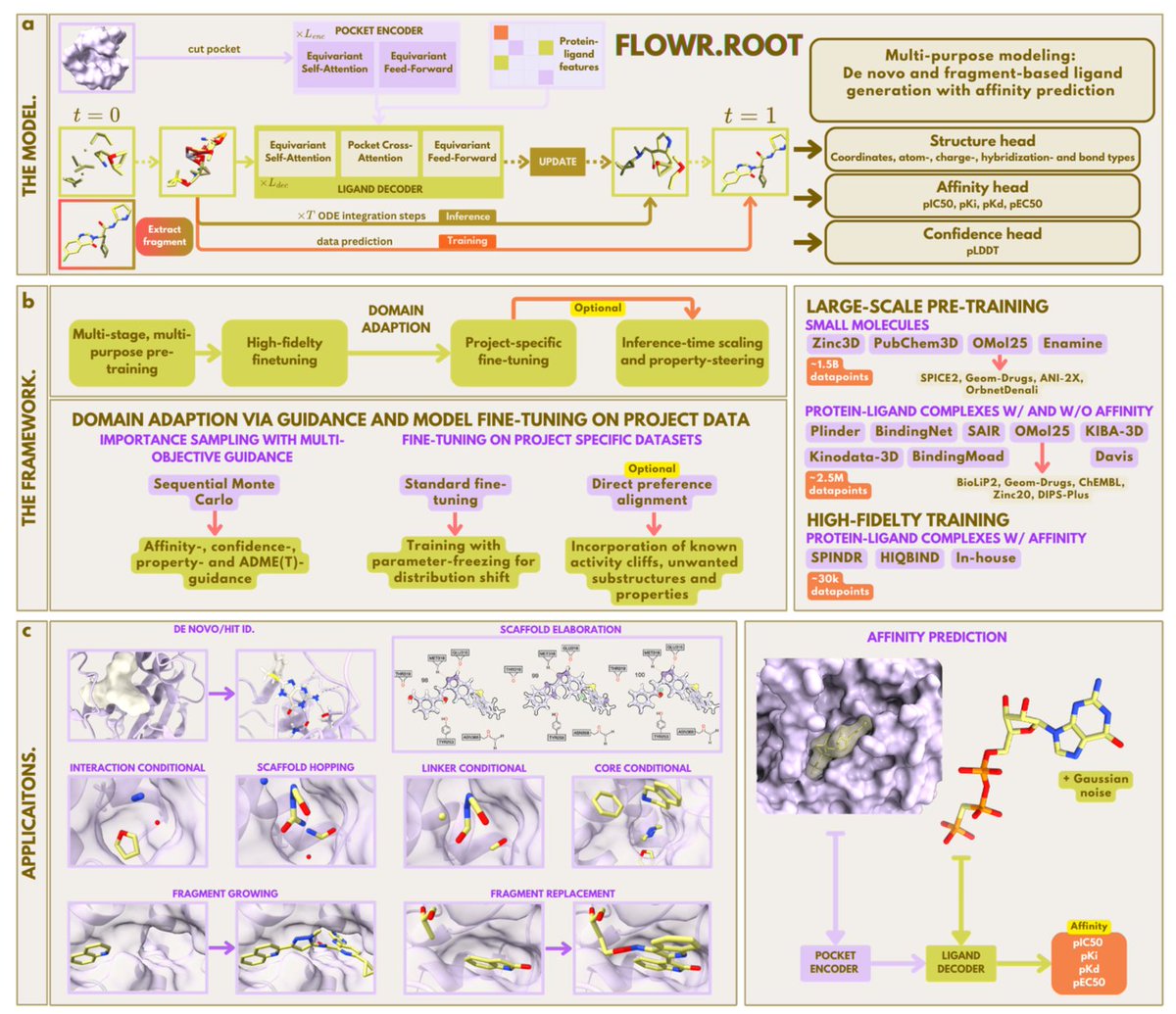
Flowr.Root – A Flow Matching Based Foundation Model for Joint Multi-Purpose Structure-Aware 3D Ligand Generation and Affinity Prediction
1. A new SE(3)-equivariant flow‑matching architecture learns to place realistic 3D ligands directly inside protein pockets, while simultaneously predicting binding potency for pIC50, pKi, pKd, and pEC50, and estimating confidence via a pLDDT head.
2. The model unifies multiple design modalities—de novo synthesis, interaction‑ or pharmacophore‑conditional sampling, scaffold hopping, and fragment growing/replacement—within a single backbone, making it a versatile tool from hit identification to lead optimization.
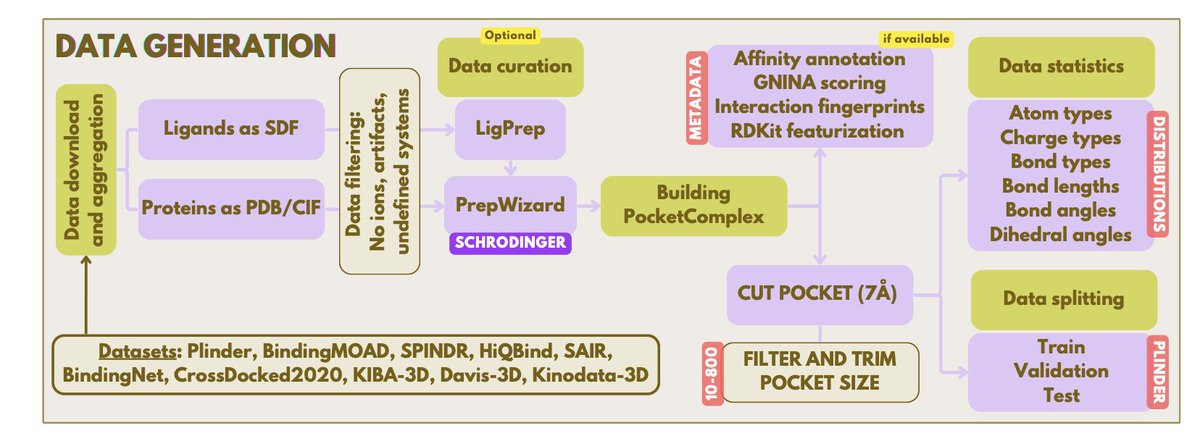
3. Training follows a three‑stage pipeline: (i) massive pre‑training on 1.5 B small‑molecule conformations and 2.5 M mixed‑fidelity protein‑ligand complexes; (ii) refinement on curated, high‑quality co‑crystal data (SPINDR, HIQBIND); (iii) rapid, parameter‑efficient LoRA fine‑tuning for project‑specific SAR alignment.
4. On standard benchmarks, FLOWR.ROOT surpasses diffusion‑based baselines with a PoseBusters‑validity of 0.97, strain energy 50 kcal/mol, and docking scores –7.5 kcal/mol, while delivering leading affinity accuracy (Pearson 0.86, RMSE 0.93 kcal/mol on the FEP /OpenFE benchmark) and 200× speed gains over physics‑based methods.
5. The affinity head predicts each potency endpoint separately, achieving Pearson 0.92 for pIC50 and 0.76 for pKi on HIQBIND, with a confidence estimator that flags low‑confidence poses.
6. Inference‑time importance sampling steers sampling toward higher affinity without retraining, improving predicted pIC50 distributions and maintaining geometric fidelity, as shown on the HIQBIND dataset.
7. Case studies demonstrate real‑world impact: joint optimization of CK2α vs CLK3 achieves selective ligands with stronger predicted affinity to CK2α, and quantum‑mechanical validation on TYK2, ERα, and BACE1 confirms strong correlation between predicted and calculated binding energies.
8. LoRA fine‑tuning transforms zero‑shot predictions on proprietary datasets and the PDE10A benchmark from poor correlations to R² ≈ 0.73 and Spearman ≈ 0.97, underscoring the power of efficient domain adaptation.
9. FLOWR.ROOT therefore offers a comprehensive, adaptable foundation for structure‑based drug design, integrating geometry‑aware generation, multi‑endpoint affinity prediction, confidence estimation, property‑guided sampling, and rapid project‑specific calibration.
📜 Paper: arxiv.org/abs/2510.02578
#DrugDiscovery #ComputationalChemistry #MachineLearning #DeepLearning #SBDD #LigandDesign #AffinityPrediction #GenerativeModels #SE3Equivariance
6
52
2,247

9 Oct 2025
Flowr.root – A Flow Matching Based Foundation Model for Joint Multi-Purpose Structure-Aware 3D Ligand Generation and Affinity Prediction
1. Flowr.root is a novel SE(3)-equivariant flow-matching model that revolutionizes 3D ligand generation and binding affinity prediction. It integrates pocket-aware ligand design with multi-endpoint affinity prediction, offering a unified framework for structure-based drug design.
2. The model supports multiple design modes, including de novo generation, interaction/pharmacophore-conditional sampling, fragment elaboration, and multi-endpoint affinity prediction. This versatility makes it suitable for various stages of drug discovery, from hit identification to lead optimization.
3. Flowr.root achieves state-of-the-art performance in both unconditional 3D molecule and pocket-conditional ligand generation. It produces geometrically realistic, low-strain structures with high computational efficiency, outperforming recent models on established benchmark datasets.
4. The integrated affinity prediction module demonstrates superior accuracy on the SPINDR test set and outperforms recent models on the Schrödinger FEP /OpenFE benchmark, while offering substantial speed advantages.
5. As a foundation model, Flowr.root requires continuous parameter-efficient fine-tuning on project-specific datasets to account for unseen structure-activity landscapes. This approach yields strong correlation with experimental in-house data, making it a dynamic companion for early-stage drug discovery campaigns.
6. The model’s joint generation and affinity prediction capabilities enable inference-time scaling through importance sampling, effectively steering molecular design toward higher-affinity compounds. Case studies validate this approach, showing significant correlation between predicted and quantum-mechanical binding energies.
7. By integrating structure-aware generation, affinity estimation, and property-guided sampling within a unified framework, Flowr.root provides a comprehensive foundation for structure-based drug design, spanning hit identification through lead optimization.
📜Paper: arxiv.org/abs/2510.02578
#FlowrRoot #DrugDiscovery #StructureBasedDesign #LigandGeneration #AffinityPrediction
1
4
1,158
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
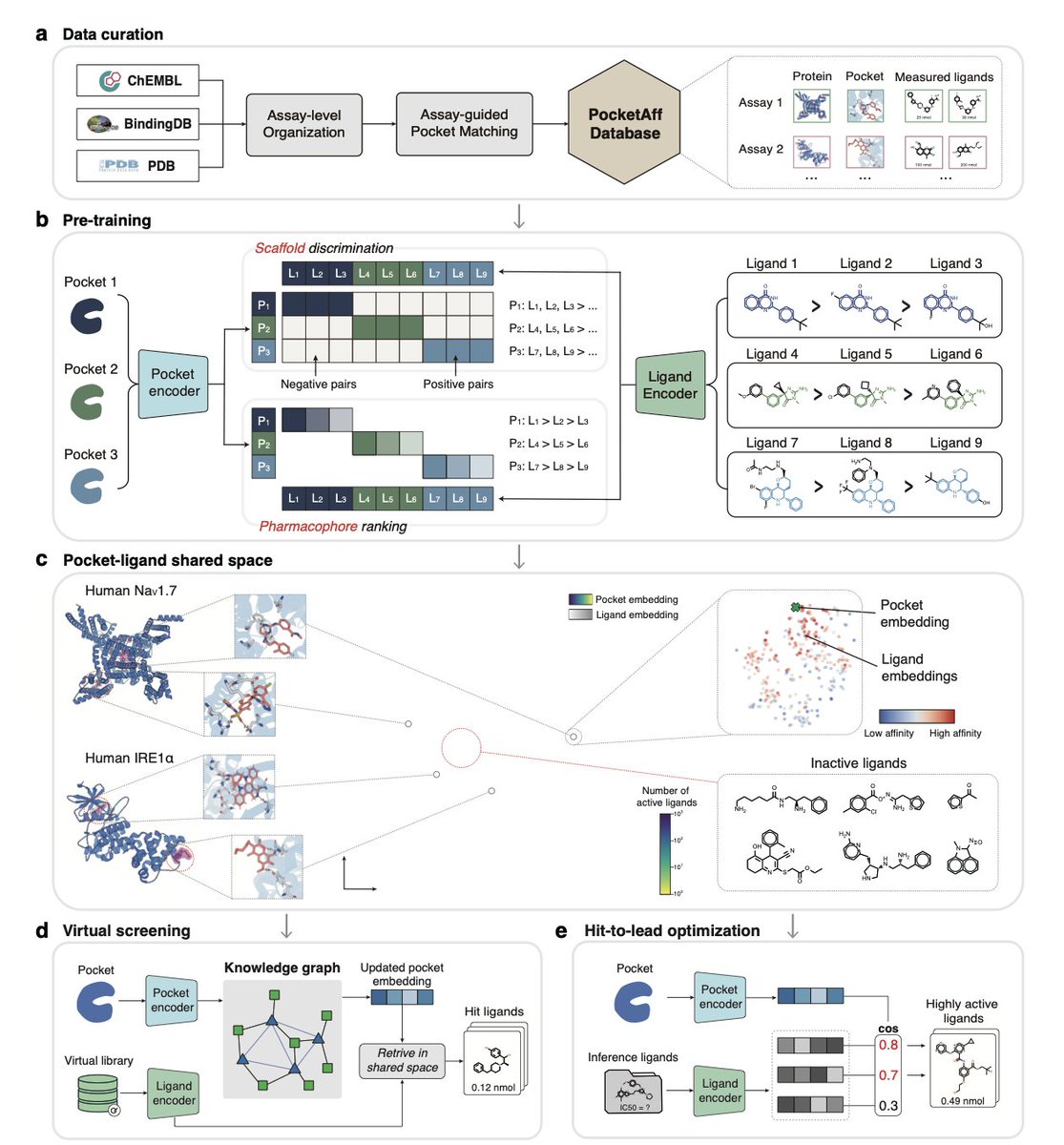
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
11
1,049
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
4
32
1,810
27 May 2025
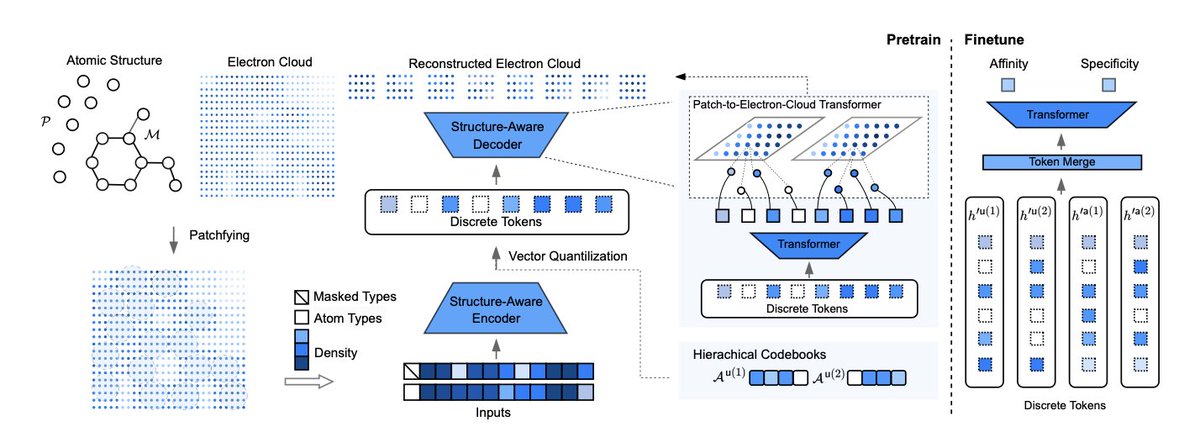
Tokenizing Electron Cloud in Protein-Ligand Interaction Learning
1.This paper presents ECBind, a novel framework that tokenizes electron cloud signals into quantized embeddings to enhance protein-ligand interaction prediction, uncovering binding patterns beyond atomic structure and achieving state-of-the-art results across multiple benchmarks.
2.Unlike conventional models that rely on atoms or fragments, ECBind explicitly incorporates electron density information—capturing non-covalent interactions, charge polarization, and π-π stacking—to provide a more physically grounded understanding of binding affinity and specificity.
3.A structure-aware transformer and hierarchical vector quantization are used to compress dense electron cloud data into discrete tokens, which are then used as inputs for downstream tasks such as binding affinity regression and activity classification.
4.ECBind is trained using a pretrain-finetune paradigm. During pretraining, the model reconstructs electron cloud signals and predicts masked atom types, enabling joint learning of 3D electronic and 2D atomic structures.
5.To address the high computational cost of computing electron clouds, the authors introduce ECBind-stdt, a student model trained via knowledge distillation from the full ECBind model, achieving nearly identical performance without requiring electron cloud inputs at inference.
6.On the MISATO dataset for relative binding affinity, ECBind-ptn achieves a 6.42% improvement in per-structure Pearson correlation and 15.58% in Spearman correlation over the best baseline, significantly outperforming fragment-level, atom-level, and hierarchical GNNs.
7.ECBind’s performance on LBA and LEP datasets also tops existing baselines. While the performance gain on LBA is more modest, this is attributed to LBA’s noisier electron cloud data, emphasizing the importance of data quality for quantum-scale modeling.
8.An ablation analysis shows that electron cloud tokenization is especially valuable for distinguishing subtle differences in ligand binding to the same protein, improving per-structure metrics by avoiding encoder over-smoothing.
9.By combining atomic and electronic structure representations, ECBind learns complementary features that enhance its capacity to model protein-ligand interactions holistically, especially in complex or "hard" binding sites.
10.The electron-aware encoder architecture is adapted from ESM3’s GeoMHA, enabling message passing that accounts for both geometric and invariant attention, and ensuring trans-rotational invariance in the learned embeddings.
11.Electron cloud patches are assigned to atoms via probabilistic sampling and neighborhood search, and are compressed into hierarchical codebooks for 2D atomic and 3D electronic attributes—enabling efficient, multi-scale representation learning.
12.ECBind sets a new direction in computational drug discovery by integrating quantum-level signals into deep learning pipelines without compromising scalability, enabling more accurate affinity predictions and ligand prioritization.
💻Code:
github.com/haitaoLin-git/ECB…
📜Paper:
arxiv.org/abs/2505.19014
#ProteinLigandBinding #QuantumML #ElectronDensity #ECBind #DrugDiscovery #AffinityPrediction #GraphNeuralNetworks #StructureBasedDesign #MolecularRepresentation #Bioinformatics
1
2
25
2,609
21 Apr 2025
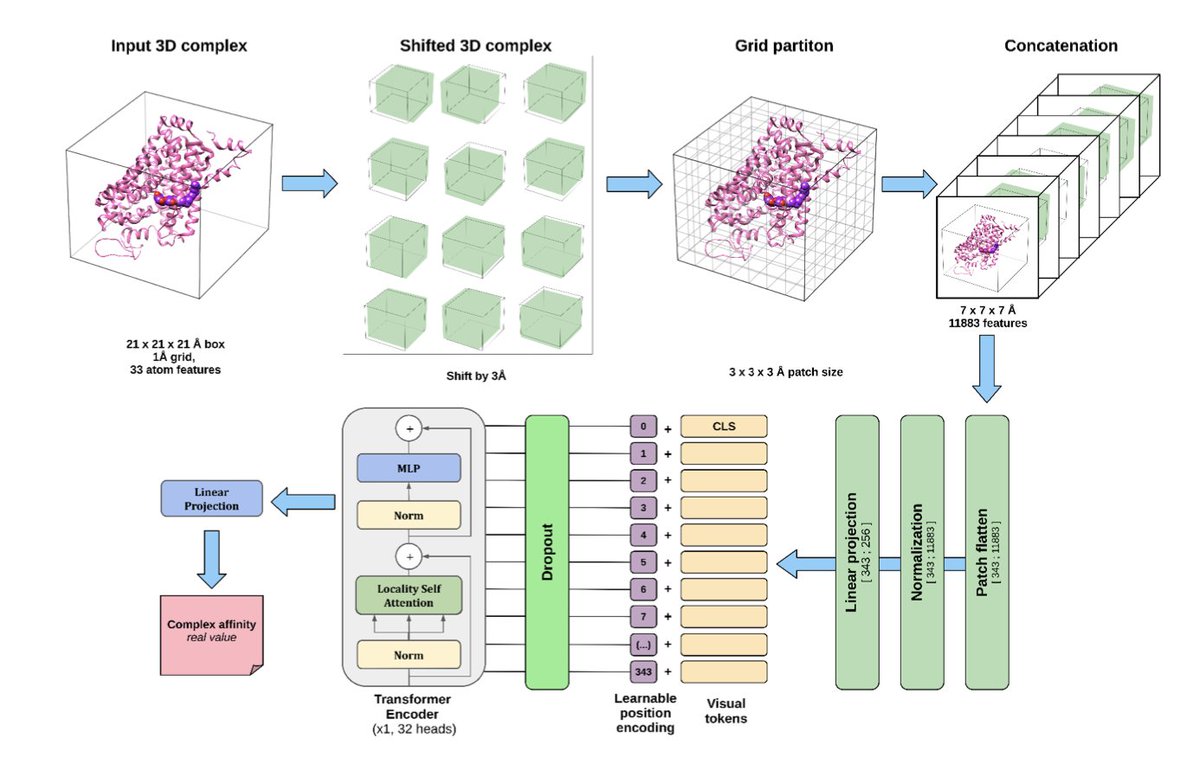
Application of vision transformers to protein-ligand affinity prediction
1. This study explores the use of Vision Transformers (ViTs) to predict protein-ligand binding affinity from 3D structural data, achieving state-of-the-art performance on challenging benchmarks like CoreSet_2013 and CASF_2016.
2. The ViT model processes voxelized 3D grids of protein-ligand complexes using shifted patch tokenization and learns rich spatial representations without relying on prior geometric assumptions—capturing long-range dependencies critical to binding affinity.
3. On CoreSet_2013, ViT achieved the lowest RMSE (1.332) among all compared methods and demonstrated strong generalization with minimal performance drop between CASF_2016 and CoreSet_2013, indicating robust behavior across datasets.
4. Ablation studies reveal that pharmacophore features are crucial for accurate prediction, while protein secondary structure and amino acid types contribute more modestly, suggesting atomic-level features alone can support high model performance.
5. Learnable positional encoding significantly boosts accuracy over 1D/3D sinusoidal or absent encodings, enabling the model to better exploit spatial organization in 3D grids—essential for reasoning about molecular interactions.
6. Rotational data augmentation dramatically improves performance, especially with 64 transformations, indicating that ViT models benefit heavily from orientation diversity to overcome dataset sparsity and overfitting.
7. Explainability analysis shows the model focuses attention on ligand-centered patches and spatially proximal interaction zones, with the [CLS] token attention peaking at the binding site—confirming biologically meaningful learning.
8. Compared to other methods like OnionNet-2, SS-GNN, CAPLA, and Pafnucy, ViT outperforms most CNN- and ML-based baselines on RMSE and Pearson correlation for affinity prediction, validating the transformer-based approach in 3D molecular settings.
9. However, conformational sensitivity remains a challenge: on MD-augmented structures, ViT predictions vary substantially, similar to other neural network models—highlighting the need for robustness training using diverse conformers.
10. The architecture combines shifted grids, locality-aware attention (LSA), and high-dimensional learned patch embeddings, effectively handling sparse volumetric input typical of protein-ligand complexes.
11. Overall, the ViT model provides a scalable and interpretable framework for structure-based affinity prediction and may generalize to other biomolecular interaction tasks like protein-protein or nucleic acid binding.
💻Code: github.com/JPoziemski/VIT_fo…
📜Paper: doi.org/10.26434/chemrxiv-20…
#DrugDiscovery #ProteinLigand #VisionTransformers #AffinityPrediction #ComputationalBiology #AI4Science #MolecularModeling #ViT
1
17
63
5,601
21 Apr 2025
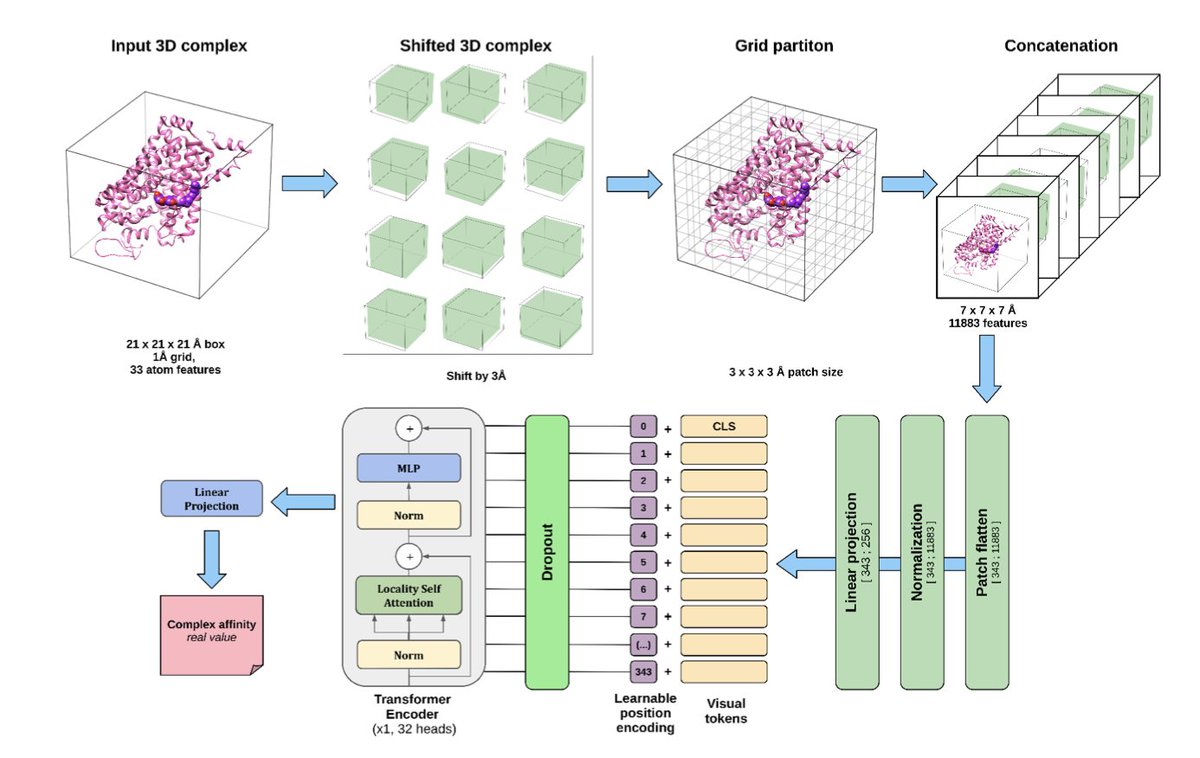
Application of vision transformers to protein-ligand affinity prediction
1. This study explores the use of Vision Transformers (ViTs) to predict protein-ligand binding affinity from 3D structural data, achieving state-of-the-art performance on challenging benchmarks like CoreSet_2013 and CASF_2016.
2. The ViT model processes voxelized 3D grids of protein-ligand complexes using shifted patch tokenization and learns rich spatial representations without relying on prior geometric assumptions—capturing long-range dependencies critical to binding affinity.
3. On CoreSet_2013, ViT achieved the lowest RMSE (1.332) among all compared methods and demonstrated strong generalization with minimal performance drop between CASF_2016 and CoreSet_2013, indicating robust behavior across datasets.
4. Ablation studies reveal that pharmacophore features are crucial for accurate prediction, while protein secondary structure and amino acid types contribute more modestly, suggesting atomic-level features alone can support high model performance.
5. Learnable positional encoding significantly boosts accuracy over 1D/3D sinusoidal or absent encodings, enabling the model to better exploit spatial organization in 3D grids—essential for reasoning about molecular interactions.
6. Rotational data augmentation dramatically improves performance, especially with 64 transformations, indicating that ViT models benefit heavily from orientation diversity to overcome dataset sparsity and overfitting.
7. Explainability analysis shows the model focuses attention on ligand-centered patches and spatially proximal interaction zones, with the [CLS] token attention peaking at the binding site—confirming biologically meaningful learning.
8. Compared to other methods like OnionNet-2, SS-GNN, CAPLA, and Pafnucy, ViT outperforms most CNN- and ML-based baselines on RMSE and Pearson correlation for affinity prediction, validating the transformer-based approach in 3D molecular settings.
9. However, conformational sensitivity remains a challenge: on MD-augmented structures, ViT predictions vary substantially, similar to other neural network models—highlighting the need for robustness training using diverse conformers.
10. The architecture combines shifted grids, locality-aware attention (LSA), and high-dimensional learned patch embeddings, effectively handling sparse volumetric input typical of protein-ligand complexes.
11. Overall, the ViT model provides a scalable and interpretable framework for structure-based affinity prediction and may generalize to other biomolecular interaction tasks like protein-protein or nucleic acid binding.
💻Code: github.com/JPoziemski/VIT_fo…
📜Paper: doi.org/10.26434/chemrxiv-20…
#DrugDiscovery #ProteinLigand #VisionTransformers #AffinityPrediction #ComputationalBiology #AI4Science #MolecularModeling #ViT
5
17
1,869