
22 Aug 2025
Learning Protein-Ligand Binding in Hyperbolic Space
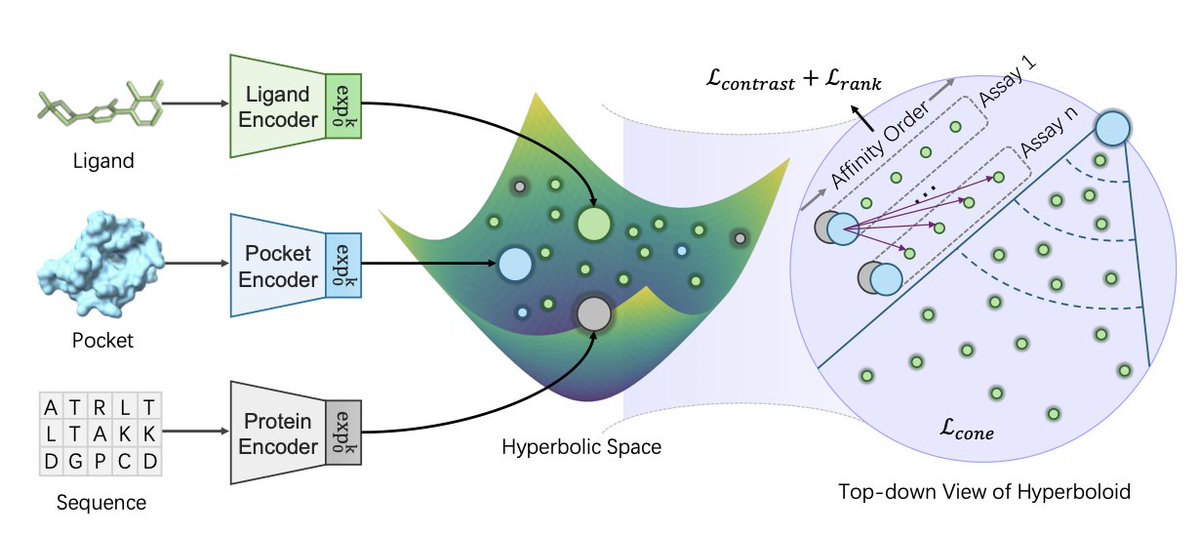
1. HypSeek, a new hyperbolic representation learning framework, improves virtual screening and affinity ranking for drug discovery by embedding ligands, protein pockets, and sequences into Lorentz-model hyperbolic space. It leverages the space's exponential geometry and negative curvature to better model molecular interactions.
2. The model shows significant improvements on benchmarks, increasing early enrichment in virtual screening on DUD-E from 42.63% to 51.44% and affinity ranking correlation on JACS from 0.5774 to 0.7239.
3. A key strength of HypSeek is its ability to handle "activity cliffs," cases where structurally similar ligands have very different binding affinities. The hyperbolic space can amplify functional differences even when structural similarities are tight, a task where Euclidean embeddings struggle.
4. The theoretical analysis confirms that hyperbolic geometry is better for separating these challenging ligand pairs. Euclidean distance grows linearly, while hyperbolic distance grows exponentially, allowing for greater separation of functionally different but structurally similar molecules.
5. The framework unifies both virtual screening and affinity ranking tasks and introduces a protein-guided three-tower architecture to enhance representational structure.
6. A comparison with the state-of-the-art LigUnity model on activity cliff pairs shows that HypSeek's hyperbolic score differences are an order of magnitude larger than Euclidean score differences, providing a clearer distinction between ligands.
7. Even in cases where free energy perturbation (FEP) predicts the wrong direction of affinity change, the hyperbolic score still aligns with the experimental ordering.
8. The paper provides a detailed theoretical motivation for why hyperbolic space is better suited for this problem, demonstrating how small angular differences in the hyperbolic embedding can lead to large separations.
9. The authors also outline the metrics used for virtual screening and affinity ranking, including AUROC, BEDROC80.5, Enrichment Factor, and Pearson and Spearman correlations.
10. This work highlights the potential of hyperbolic geometry as a powerful inductive bias for protein-ligand modeling, offering a more expressive and affinity-sensitive approach.
📜Paper: arxiv.org/abs/2508.15480
#DrugDiscovery #AIinDrugDiscovery #ComputationalBiology #MachineLearning #HyperbolicGeometry #ProteinLigandBinding #VirtualScreening #AffinityRanking
1
18
109
5,444
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
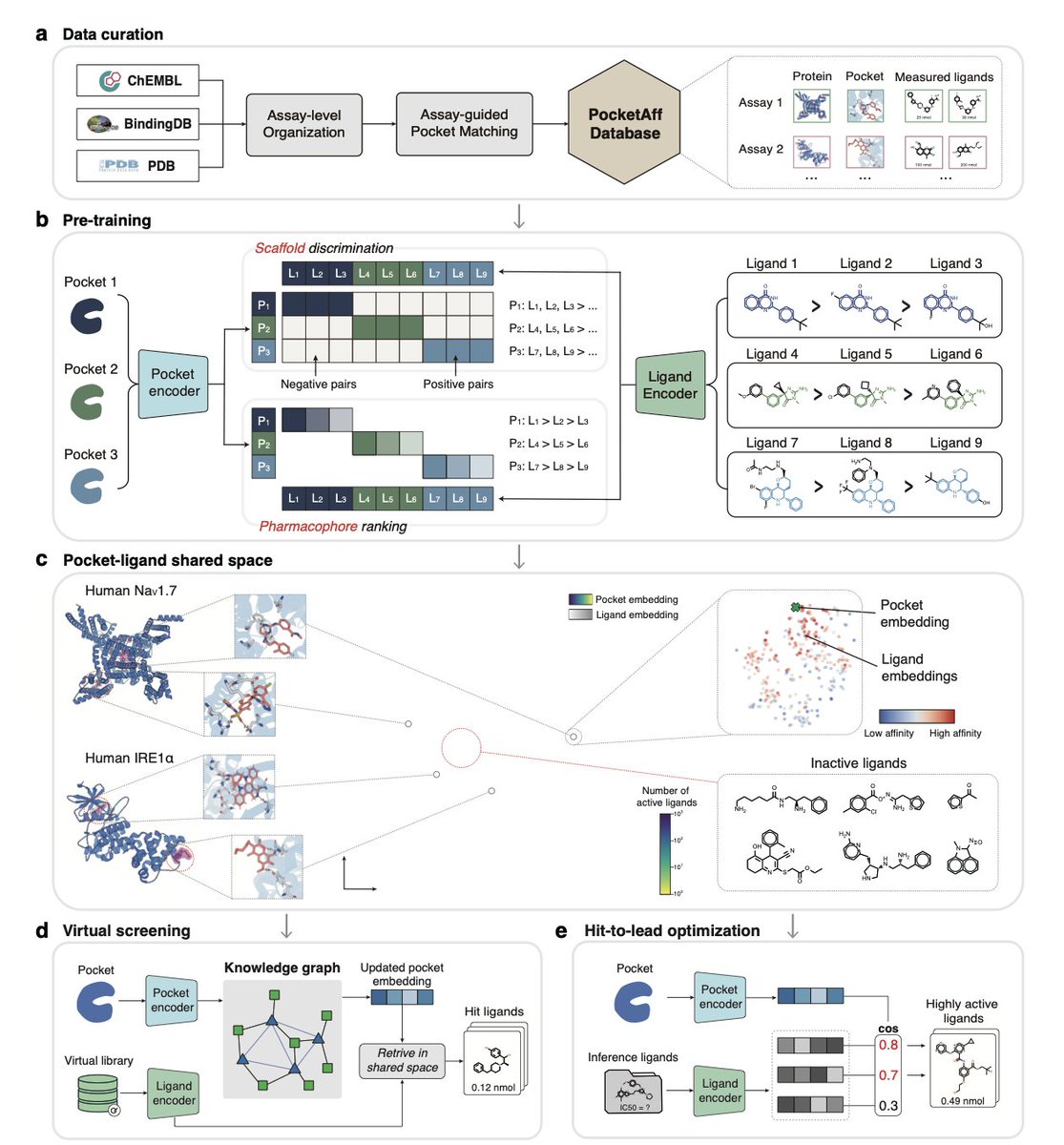
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
11
1,049
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
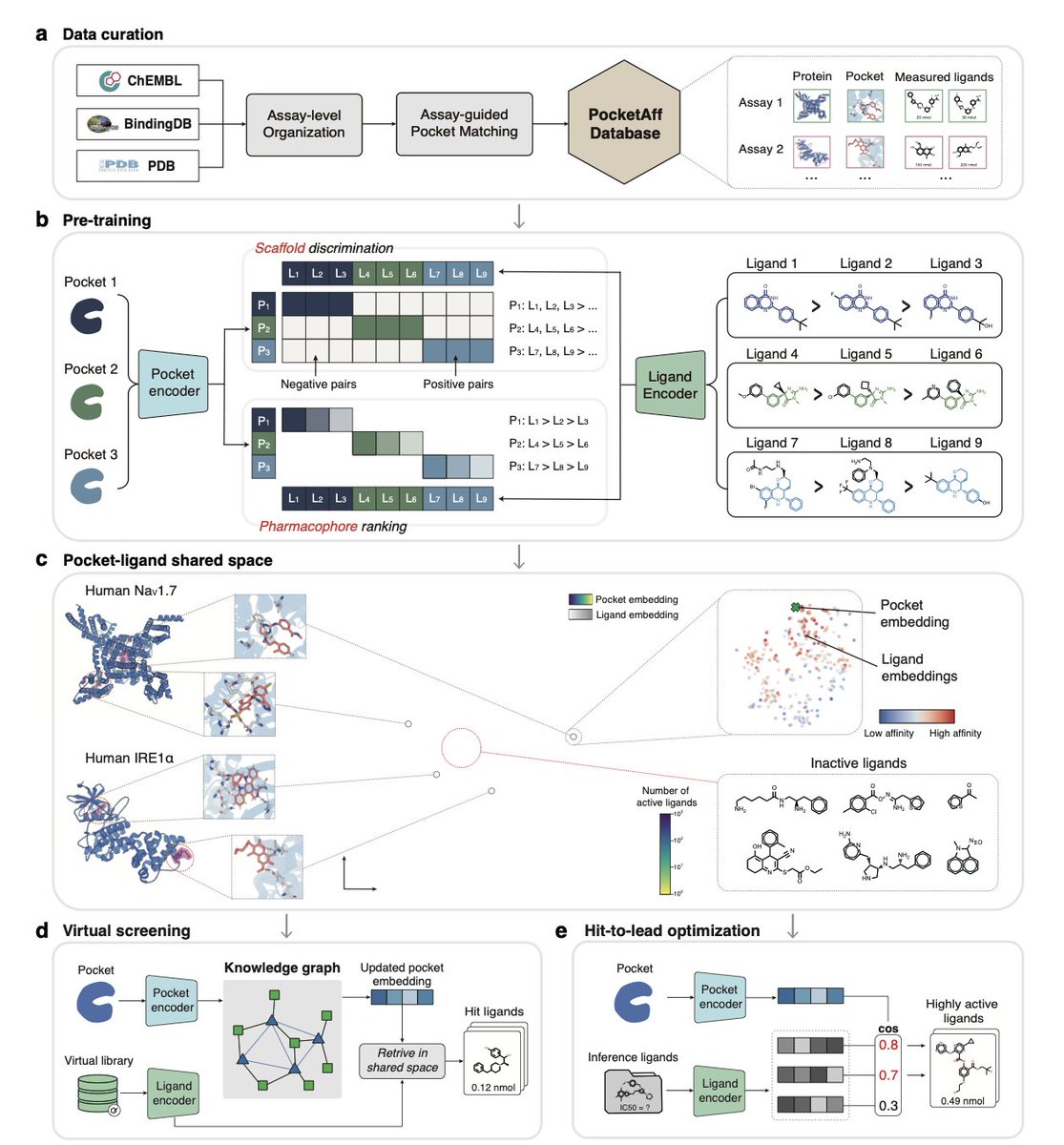
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
4
32
1,810

25 Feb 2025
LigUnity unifies virtual screening & hit-to-lead optimization.
A single foundation model that slashes FEP costs and shows some generalizability to novel targets.
I'm wondering if we're looking at a future where AI-driven affinity predictors become the new standard 👀
1
2
221
23 Feb 2025
A foundation model for protein-ligand affinity prediction through jointly optimizing virtual screening and hit-to-lead optimization
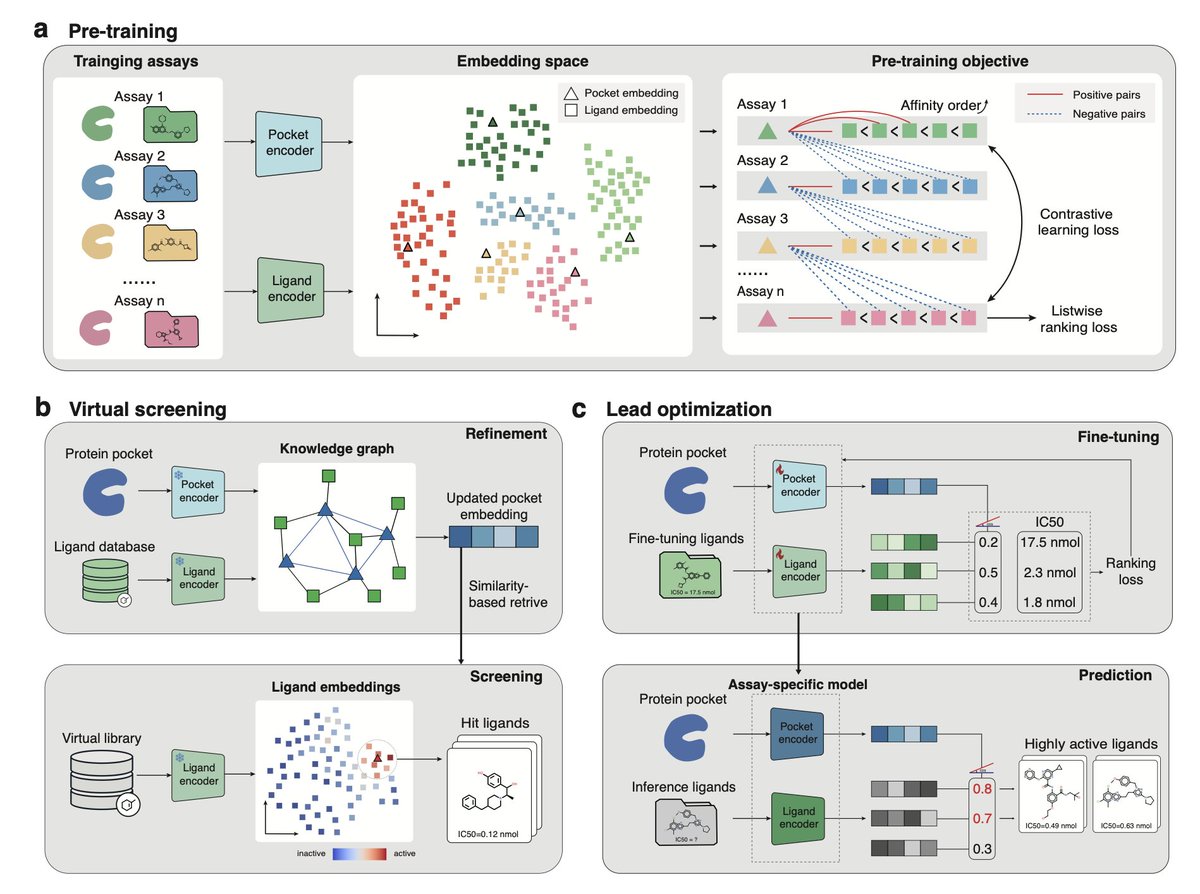
1. LigUnity is a novel foundation model that jointly optimizes protein-ligand virtual screening and hit-to-lead optimization, leveraging the synergy between these two tasks to enhance the overall drug discovery pipeline.
2. The model uses contrastive learning to distinguish between active and inactive ligands during virtual screening and a listwise ranking approach to optimize the affinity prediction for hit-to-lead optimization, improving performance across various benchmarks.
3. LigUnity outperforms 24 competing methods in virtual screening tasks on several benchmarks, such as DUD-E, Dekois 2.0, and LIT-PCBA, with significant improvements in the enrichment factor and faster screening speed.
4. The model also excels in hit-to-lead optimization tasks, achieving superior performance compared to traditional computational methods like free energy perturbation (FEP), particularly in zero-shot and few-shot settings.
5. The integration of LigUnity in an active learning framework shows its ability to identify optimal binding ligands for TYK2, a therapeutic target for autoimmune diseases, achieving over 40% improved prediction performance.
6. LigUnity's versatility is highlighted through its application to diverse settings, including split-by-time, split-by-scaffold, and split-by-unit settings, where it consistently outperforms other methods and generalizes well to unseen proteins and novel chemical scaffolds.
7. The ability to handle different assay types, including those using percentage units and real-world datasets, makes LigUnity an ideal tool for drug discovery, offering significant improvements over existing methods in both speed and accuracy.
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #Bioinformatics #AIinPharma #ComputationalChemistry #ProteinLigandAffinity #DeepLearning #HitToLead #ActiveLearning #ProteinModeling #LigandOptimization
1
7
58
4,751
23 Feb 2025
A foundation model for protein-ligand affinity prediction through jointly optimizing virtual screening and hit-to-lead optimization
1. LigUnity is a novel foundation model that jointly optimizes protein-ligand virtual screening and hit-to-lead optimization, leveraging the synergy between these two tasks to enhance the overall drug discovery pipeline.
2. The model uses contrastive learning to distinguish between active and inactive ligands during virtual screening and a listwise ranking approach to optimize the affinity prediction for hit-to-lead optimization, improving performance across various benchmarks.
3. LigUnity outperforms 24 competing methods in virtual screening tasks on several benchmarks, such as DUD-E, Dekois 2.0, and LIT-PCBA, with significant improvements in the enrichment factor and faster screening speed.
4. The model also excels in hit-to-lead optimization tasks, achieving superior performance compared to traditional computational methods like free energy perturbation (FEP), particularly in zero-shot and few-shot settings.
5. The integration of LigUnity in an active learning framework shows its ability to identify optimal binding ligands for TYK2, a therapeutic target for autoimmune diseases, achieving over 40% improved prediction performance.
6. LigUnity's versatility is highlighted through its application to diverse settings, including split-by-time, split-by-scaffold, and split-by-unit settings, where it consistently outperforms other methods and generalizes well to unseen proteins and novel chemical scaffolds.
7. The ability to handle different assay types, including those using percentage units and real-world datasets, makes LigUnity an ideal tool for drug discovery, offering significant improvements over existing methods in both speed and accuracy.
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #Bioinformatics #AIinPharma #ComputationalChemistry #ProteinLigandAffinity #DeepLearning #HitToLead #ActiveLearning #ProteinModeling #LigandOptimization

4
36
1,865