SMDD-Bench: Can LLMs Solve Real-World Small Molecule Drug Design Tasks?
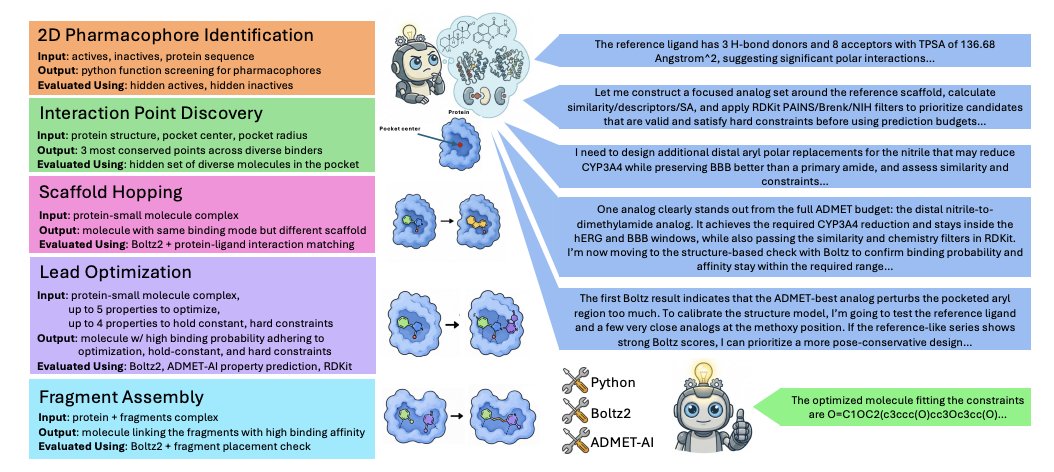
1. The paper introduces SMDD-Bench, an agentic, multi-turn, long-horizon benchmark for evaluating LLM agents on realistic small-molecule drug design workflows, moving beyond single-turn chemistry QA and toy tasks.
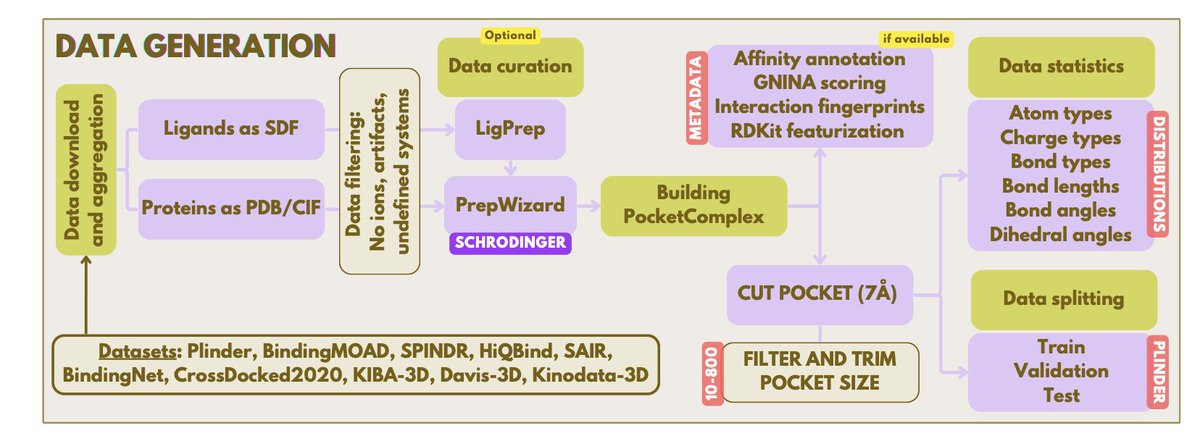
2. SMDD-Bench contains 502 guaranteed-solvable task instances spanning 5 task types: 2D Pharmacophore Identification (25), Interaction Point Discovery (25), Scaffold Hopping (52), Lead Optimization (340), and Fragment Assembly (60), covering 102 unique protein targets and broad chemical space.
3. A key methodological contribution is “witness-aware task generation”: for task types where solvability is not guaranteed by default (Scaffold Hopping, Lead Optimization, Fragment Assembly), each instance is constructed together with a hidden witness molecule that is known to satisfy the evaluation criteria, ensuring every task is solvable by construction without human curation.
4. The benchmark is designed to test capabilities needed for practical computational medicinal chemistry: chemical/biological reasoning, 3D geometric intuition, planning under limited expensive oracle calls, and tool use (Python/RDKit workflows, structure/interaction analysis), rather than only knowledge recall.
5. Evaluation is fully automated (no human grading) and uses a tool stack typical of computational drug design pipelines: RDKit for chemistry and filters, PLIP for interaction fingerprints, OpenBabel utilities, Boltz2 for protein–ligand co-folding plus binding probability/affinity, and ADMET-AI for multi-property prediction.
6. Each task type targets a distinct real-world skill: (a) 2D Pharmacophore ID requires writing a Python function that generalizes from 10 actives 10 inactives to hidden ChEMBL actives/inactives; (b) Interaction Point Discovery requires predicting 3 conserved pocket “hotspots” (3D coordinates type) derived from large co-crystal ensembles; (c) Scaffold Hopping requires low 2D similarity but matching 3D interaction patterns; (d) Lead Optimization requires multi-objective improvement while holding other properties constant under hard constraints; (e) Fragment Assembly requires linking 1–2 posed fragments while preserving pose and binding.
7. Benchmarking 7 frontier LLMs with a minimalist ReAct-style agent (no internet, obfuscated IDs, limited oracle budgets: 8 Boltz2 calls 15 ADMET-AI calls) shows substantial headroom: the best model (GPT-5.4) solves 40.2% overall, with most wins coming from Lead Optimization rather than 3D-heavy tasks.
8. Results highlight a consistent weakness in 3D reasoning: Interaction Point Discovery is near-zero for most models, and Scaffold Hopping / Fragment Assembly success rates are also very low, suggesting that “tool access” alone does not yield reliable 3D pocket/pose reasoning.
9. The authors analyze novelty and diversity of generated molecules. Many submitted molecules are “novel” relative to major databases (ChEMBL, SureChEMBL, PubChem, BindingDB), but repeated runs reveal limited diversity: agents often converge to similar solutions (high pairwise Tanimoto among successful outputs), which is misaligned with real lead-optimization needs where multiple diverse viable candidates are preferred.
10. An “enumeration vs. selection” study suggests agents can mention many candidate SMILES that would pass evaluation but fail to choose them for oracle calls/submission—especially in Scaffold Hopping—pointing to planning/selection as a bottleneck, not only molecule generation.
11. The paper also provides a practical adoption path via SMDD-Bench Lite (100 instances) to reduce compute barriers while keeping difficulty representative, plus SMDD-Bench Diversity (20 hard lead-optimization instances, run 10x each) to quantify output diversity and novelty under repeated trials.
12. Common failure modes observed in traces include poor cross-turn SAR synthesis (not learning exclusion rules from failed candidates), incoherent multi-turn planning (re-testing or re-proposing failed molecules), and tool-specific coding errors (e.g., malformed calls, RDKit conversion issues), emphasizing that robust agent scaffolding matters as much as base-model capability.
📜Paper: arxiv.org/abs/2605.21740
#LLM #DrugDiscovery #MedicinalChemistry #Cheminformatics #Benchmark #Agents #ADMET #StructureBasedDesign #ComputationalBiology #RDKit
1
3
11
1,643
Empowering Chemical Structures with Biological Insights for Scalable Phenotypic Virtual Screening
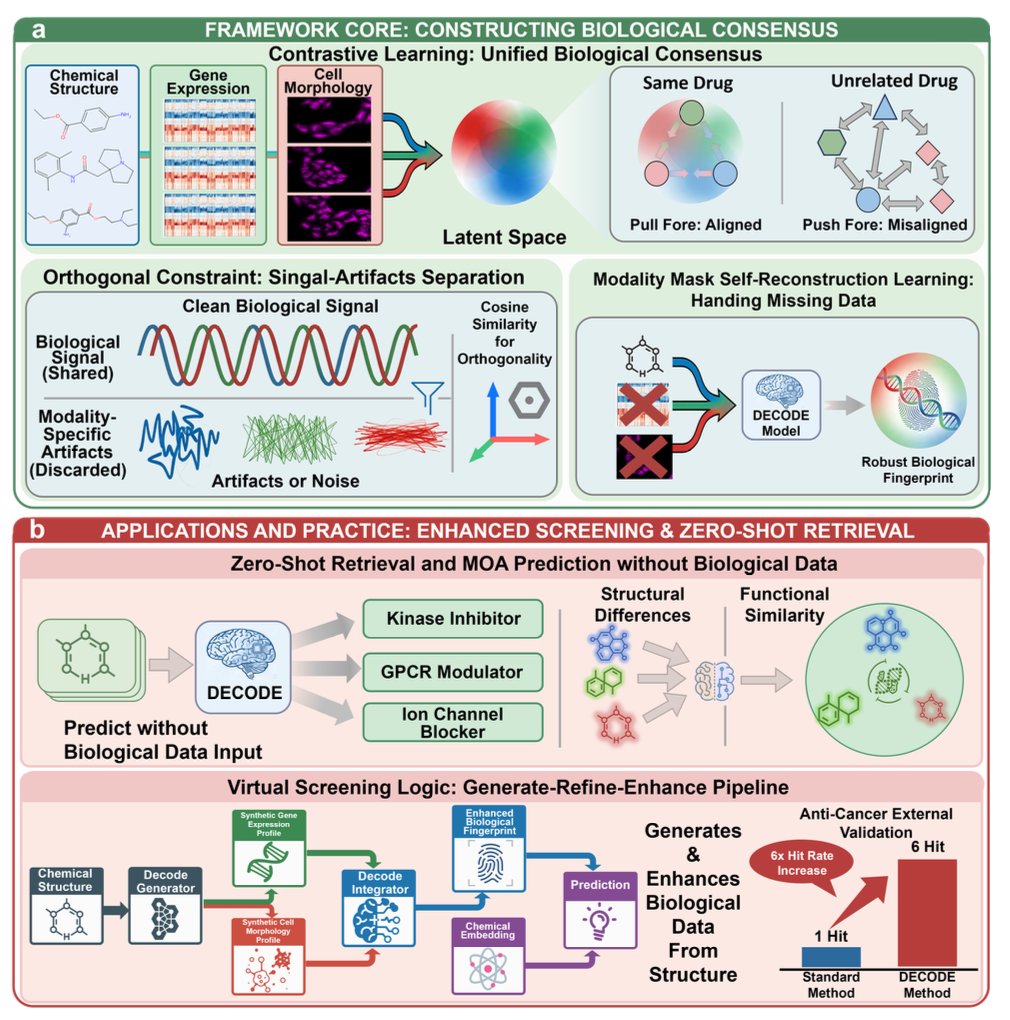
1. DECODE introduces a new paradigm that turns raw SMILES into biologically meaningful fingerprints by learning from paired transcriptomic and morphological profiles during training, yet requiring only chemical structures at inference.
2. The framework treats biological modalities as privileged information: it aligns chemical embeddings with a measurement‑invariant consensus space while ignoring assay‑specific noise, allowing the model to infer functional effects for unseen compounds.
3. A geometric disentanglement module splits each modality into a shared biological signal and an orthogonal, modality‑specific noise component, coupled with a contrastive loss that forces the chemical encoder to match the consensus, producing a robust, noise‑free fingerprint.
4. In zero‑shot drug retrieval, DECODE identifies functionally equivalent compounds with over 20 % higher top‑5 recall than traditional chemical similarity baselines, correctly clustering drugs that share mechanisms despite divergent scaffolds.
5. For sparse‑label mechanism‑of‑action classification, the method yields a 15–20 % F1‑score boost over expert MLPs, demonstrating that the consensus space filters out conflicting experimental artifacts that degrade standard fusion approaches.
6. A Generate‑Refine‑Enhance pipeline augments virtual screening: synthetic transcriptomic and morphological profiles are generated, refined, and combined with the structural encoding, achieving a six‑fold increase in hit rates for novel anti‑cancer agents compared to structure‑only models.
7. Ablation studies confirm that both the modal‑alignment phase and the orthogonality constraint are essential; removing either leads to significant drops in retrieval, MOA prediction, and hit‑rate performance.
8. Future work will embed context‑aware injection to capture tissue‑specific responses and integrate foundation models for richer biological feature extraction, further tightening the bridge between chemistry and phenotypic biology.
💻Code: github.com/lian-xiao/DECODE
📜Paper: arxiv.org/abs/2603.15006
#DrugDiscovery #Chemoinformatics #PhenotypicScreening #MachineLearning #VirtualScreening #Bioinformatics #AIinMedicine #DeepLearning #CompoundProfiling #MOAPrediction #HitRateImprovement #StructureBasedDesign #PharmaTech #ComputationalBiology #OpenSource
3
16
1,274
EvoEGF-Mol: Evolving Exponential Geodesic Flow for Structure-based Drug Design
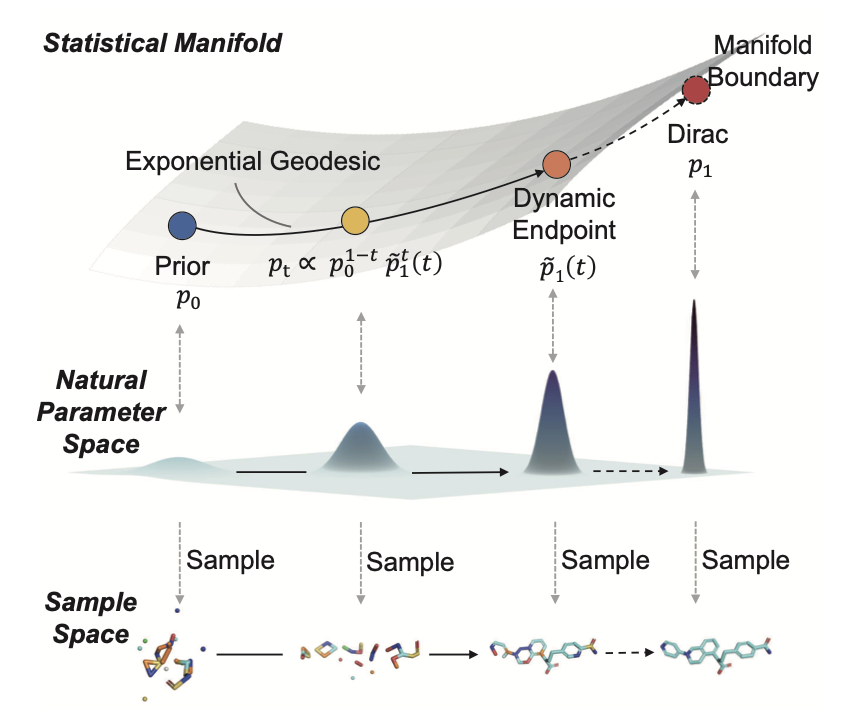
1 EvoEGF-Mol rewrites SBDD as a journey on a Fisher–Rao statistical manifold, replacing the usual “coordinates atom types” split with one composite exponential-family distribution whose natural parameters evolve along an exponential geodesic.
2 The key twist: instead of aiming at a static Dirac delta that collapses instantly, the method chases a *moving* target whose variance and concentration tighten smoothly over time, keeping the trajectory inside the manifold interior and the training signal well spread.
3 This dynamic-endpoint trick removes the “late-stage explosion” of natural parameters that cripples ordinary flow matching; the path stays numerically stable, so the network can learn fine-grained geometry–chemistry correlations all the way to t = 1.
4 Continuous atomic positions ride an isotropic Gaussian geodesic, while discrete atom and bond types travel on Dirichlet simplices; both share the same schedule, guaranteeing that 3-D coordinates and chemical identity refine together rather than drift apart.
5 On CrossDock2020 the model hits 93.4 % PoseBusters validity, lowest quartile strain energy < 9 kcal mol⁻¹, and Vina Dock scores that rival or exceed prior best methods, all with 100 % chemical sanitization and < 1 % internal clashes.
6 In the real-world MolGenBench it recovers 72 bioactive scaffolds for seen proteins and 33 for unseen ones, doubling the scaffold pass rate of the strongest baseline, although absolute hit fractions remain low, highlighting the field’s ongoing challenge.
7 Ablation shows that freezing the endpoint (naïve EGF) drops validity to 64 % and produces repulsive poses, while removing explicit bond diffusion raises strain energy by ~40 %, confirming that the coupled, evolving schedule is essential.
8 Training needs 16 h on a single GPU, inference 100 steps, and the entire framework is probability-theory-first: every update is a closed-form geodesic step on the manifold, no score-network tricks required.
💻Code: github.com/linganglab/evoeGF…
📜Paper: arxiv.org/abs/2601.22466
#DrugDiscovery #StructureBasedDesign #DiffusionModels #InformationGeometry #ExponentialFamily #AI4Science #Bioinformatics
5
28
1,550
Enabling automatic generation of protein-ligand complex datasets with atomistic detail
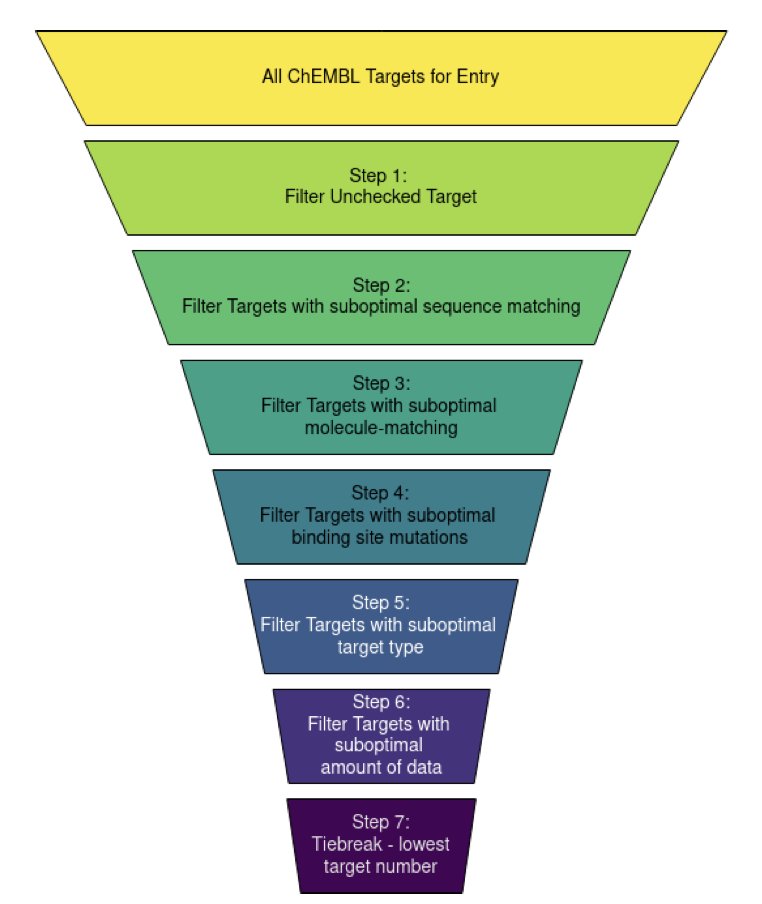
1. A new study introduces StrAcTable, an automated dataset of annotated protein-ligand complexes, combining detailed structural data from PDB and bioactivity data from ChEMBL. This dataset aims to address the challenge of linking structural and bioactivity data, which is crucial for structure-based drug design.
2. StrAcTable includes 20,063 protein-ligand complexes with bioactivity values, derived from ChEMBL Version 35. The dataset provides detailed quality assessments of the matching between protein sequences and small molecules, as well as the overall structure quality.
3. The innovation lies in the fully automated workflow, which uses tools like ActivityFinder, LigandExtractor, and StructureProfiler to integrate and assess data quality. This automation allows for sustainable growth and updates, reducing manual efforts significantly.
4. StrAcTable offers multiple versions to cater to different research needs, including filtered datasets that retain only the best-matching ChEMBL targets. This flexibility makes it a valuable resource for both machine-learning and traditional docking-scoring development.
5. The study highlights the importance of sequence and molecule matching quality, categorizing matches into Gold, Silver, and Bronze levels. This allows researchers to filter data based on desired quality criteria.
6. StrAcTable also tracks mutations in the binding site, providing insights into potential influences on binding affinity. The dataset's chemical diversity and wide range of activity values make it a comprehensive resource for drug discovery projects.
7. The authors demonstrate the potential of StrAcTable by constructing high-quality datasets for specific targets, such as the Tyrosine protein kinase ABL. This showcases how StrAcTable can be used to create tailored datasets for individual research needs.
📜Paper: biorxiv.org/content/10.64898…
💻Code: github.com/rareylab/StrAcTab…
#StrAcTable #ProteinLigandComplex #AutomatedDataset #DrugDiscovery #Bioactivity #StructureBasedDesign
2
11
1,266

25 Nov 2025
Apo2Mol: 3D Molecule Generation via Dynamic Pocket-Aware Diffusion Model
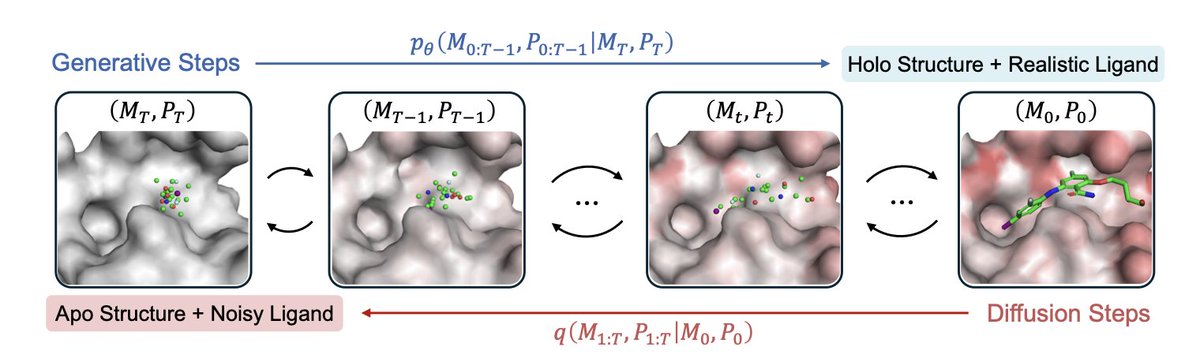
1. A new study titled "Apo2Mol" introduces a groundbreaking approach to structure-based drug design, leveraging a dynamic pocket-aware diffusion model to generate 3D molecules that account for protein flexibility. This innovation addresses a critical limitation in current methods that assume rigid protein binding pockets.
2. The core innovation of Apo2Mol lies in its ability to jointly generate ligands and their corresponding holo pocket conformations from apo states. This is achieved through a full-atom hierarchical graph-based diffusion model, which captures realistic protein-ligand interactions and conformational changes.
3. The researchers curated a high-quality dataset of over 24,000 experimentally resolved apo-holo structure pairs from the Protein Data Bank. This dataset provides a robust foundation for training the model, ensuring that it learns from real-world protein dynamics rather than relying on simulated data.
4. Empirical results demonstrate that Apo2Mol achieves state-of-the-art performance in generating high-affinity ligands. The model not only produces molecules with favorable binding affinities but also generates realistic pocket conformations, advancing the capabilities of dynamic structure-based drug design.
5. The study highlights the potential of Apo2Mol to generate novel ligands for emerging drug targets, even when only apo structures are available. This capability is crucial for practical drug discovery scenarios where holo structures are often unknown.
📜Paper: arxiv.org/abs/2511.14559v1
#DrugDesign #DiffusionModels #ProteinFlexibility #StructureBasedDesign #AIinBiology
2
15
53
3,365
19 Nov 2025
SpecLig: Energy-Guided Hierarchical Model for Target-Specific 3D Ligand Design
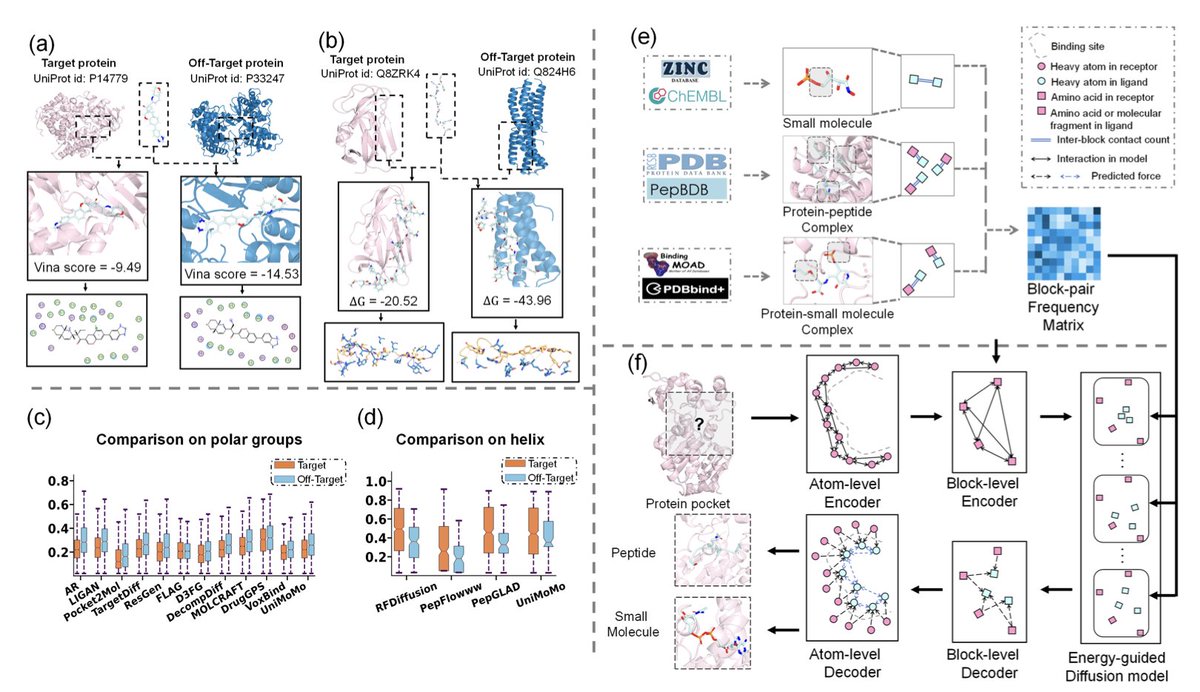
1. A new framework called SpecLig has been introduced to address the critical issue of target specificity in ligand design. This model integrates structure-based design with statistical energy guidance to generate ligands that exhibit high specificity and affinity for their intended targets.
2. SpecLig employs a hierarchical graph neural network combined with an energy-guided diffusion model. It leverages empirical block–block interaction statistics from natural protein-ligand complexes to bias the generation towards pocket-specific binding configurations.
3. The model represents protein-ligand complexes as block-based graphs, capturing both local chemistry and global topology. This hierarchical representation reduces atom-level noise and preserves fragment semantics, leading to improved specificity.
4. Evaluations on standard peptide and small molecule benchmarks show that SpecLig consistently produces ligands with higher specificity compared to existing methods, while maintaining competitive affinity and other attributes.
5. Case studies demonstrate SpecLig’s ability to mitigate off-target risks by optimizing pocket-specific geometric and chemical complementarity. This approach shows promise in advancing safer and more effective ligand candidates for drug discovery.
6. The hierarchical VAE and energy-guided diffusion components work synergistically to balance affinity and specificity. Ablation studies confirm the necessity of both components for optimal performance.
7. While SpecLig shows significant improvements in specificity, the authors note that small molecule design remains challenging due to the high complexity and sensitivity of chemical spaces. Future work may integrate richer physical cues to further enhance performance.
📜Paper: biorxiv.org/content/10.1101/…
#SpecLig #LigandDesign #TargetSpecificity #StructureBasedDesign #DrugDiscovery #AIinBiology
3
19
1,523
31 Oct 2025
CIDD: Collaborative Intelligence for Structure-Based Drug Design Empowered by LLMs
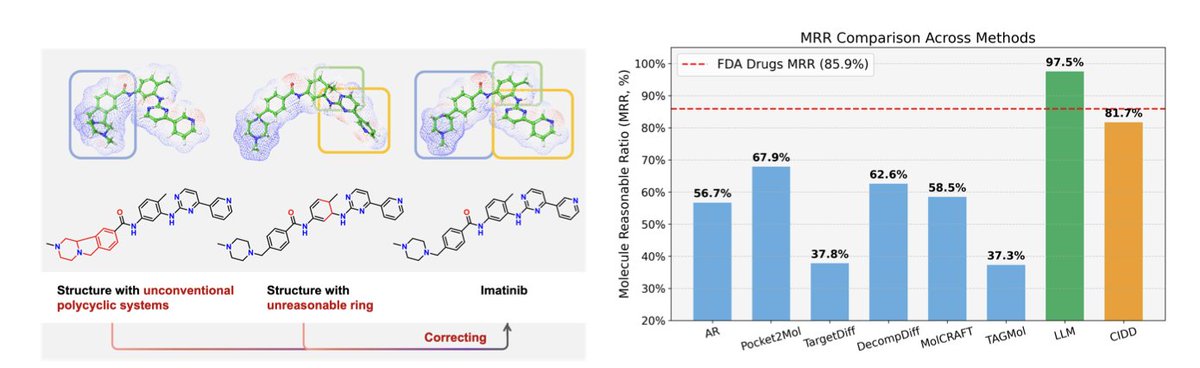
1. The article introduces CIDD, a novel framework that integrates 3D interaction modeling with the reasoning power of large language models (LLMs) to enhance structure-based drug design. This approach aims to generate molecules that are not only compatible with protein pockets but also exhibit favorable drug-likeness and structural rationality.
2. A key innovation is the introduction of the Molecule Reasonable Ratio (MRR), a metric designed to quantify the structural rationality of generated molecules. This metric reveals a significant gap between existing models and real-world approved drugs, highlighting the need for more chemically plausible molecular designs.
3. CIDD leverages LLM-based Chain-of-Thought reasoning to generate molecules. The framework includes specialized modules such as the Interaction Analysis Module, Design Module, Reflection Module, and Selection Module, each powered by LLMs, to ensure the generated molecules are both interaction-competent and structurally plausible.
4. Evaluated on the CrossDocked2020 benchmark, CIDD significantly outperforms state-of-the-art methods, improving key drug-likeness metrics like QED, SA, and MRR while maintaining competitive binding affinity. The combined success rate balancing drug-likeness and binding affinity more than doubles previous results.
5. The modular and Chain-of-Thought-guided architecture of CIDD mirrors real-world medicinal chemistry workflows, enabling interpretable and stepwise molecular design. This approach sets a foundation for future directions in rational, interpretable, and generalizable drug generation.
📜Paper: openreview.net/pdf/77cb4fc75…
#CIDD #DrugDesign #LLMs #StructureBasedDesign #MolecularGeneration
1
5
16
1,944

23 Oct 2025
📢 I am hiring a postdoc in #AI-driven #DrugDiscovery!
Join my lab in Barcelona.
Focus: AI, #Cheminformatics, #StructureBasedDesign, #CellPainting, #ImmunoOncology 💡
DM me for more information
#PostdocJobs #AI #DrugDesign #MachineLearning #Barcelona @EuroPhDNetwork @PostdocJobs
2
4
255
17 Oct 2025
Slogen: A Structure-based Lead Optimization Model Unifying Fragment Generation and Screening
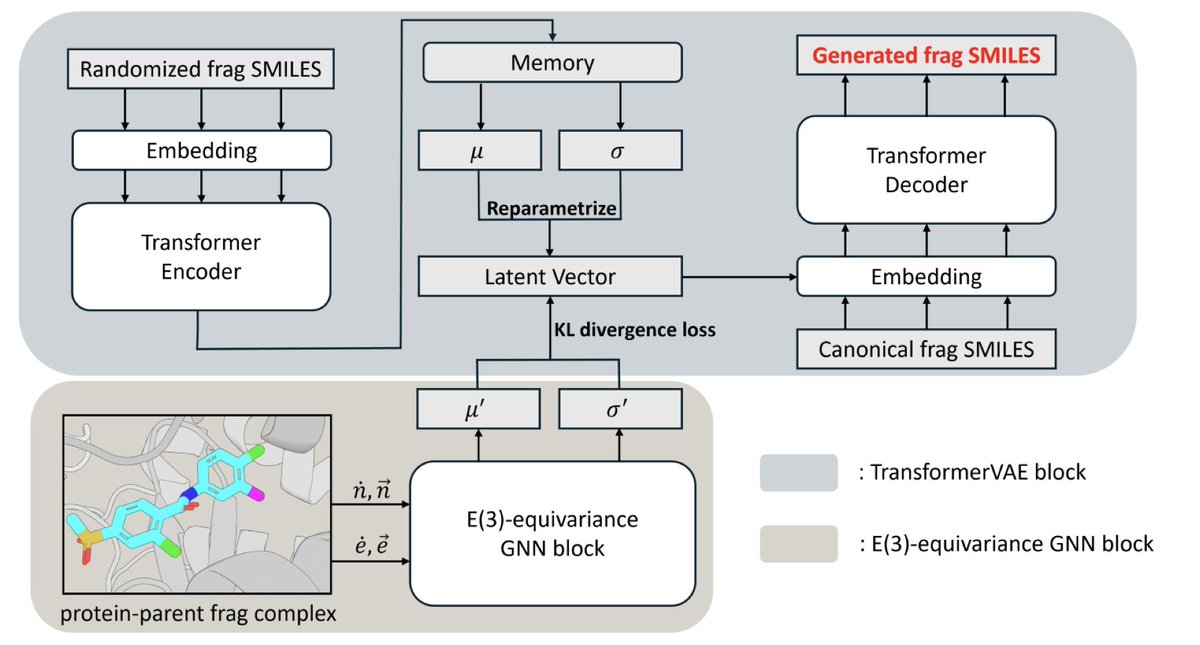
1. Slogen is a groundbreaking model in the field of structure-based drug design, aiming to enhance the efficiency and effectiveness of lead optimization. It combines fragment generation and screening in a unified framework, addressing key challenges in synthetic feasibility and structural innovation.
2. The model integrates a transformer-based variational autoencoder (VAE) with an E(3)-equivariant graph neural network (GNN). The VAE is pretrained on a diverse set of fragments, enabling both generative decoding and similarity-based screening. The GNN captures 3D protein–fragment interactions, predicting optimal fragment elaborations.
3. Slogen demonstrates superior performance in fragment elaboration tasks, achieving higher hit rates and better binding affinities compared to state-of-the-art models like Delete and DeepFrag. It also explores a broader chemical space, generating more diverse and drug-like molecules.
4. In screening tasks, Slogen shows competitive performance, particularly on the Delete test set, significantly outperforming traditional methods such as AutoDock Vina. This highlights its potential for practical applications in fragment-based drug discovery.
5. Case studies on the Smoothened receptor (SMO) and D1 dopamine receptor (D1DR) further illustrate Slogen's ability to design high-affinity, drug-like molecules. These examples demonstrate its versatility and practical utility in real-world drug design scenarios.
6. Despite its strengths, Slogen has areas for improvement. It performs well in generating chemically plausible ring structures but struggles with capturing the distribution of BM scaffolds. Future work could focus on pretraining with a more diverse fragment set and incorporating advanced GNN architectures.
7. The study emphasizes the importance of a balanced fragmentation strategy that ensures both synthetic tractability and chemical diversity. Slogen's unified approach provides a scalable route toward structure-guided lead optimization, bridging the gap between fragment generation and screening.
📜Paper: biorxiv.org/content/10.1101/…
#Slogen #LeadOptimization #StructureBasedDesign #DrugDiscovery #AIinPharma #FragmentGeneration #Screening #ComputationalBiology
2
13
1,380
9 Oct 2025
Flowr.root – A Flow Matching Based Foundation Model for Joint Multi-Purpose Structure-Aware 3D Ligand Generation and Affinity Prediction
1. Flowr.root is a novel SE(3)-equivariant flow-matching model that revolutionizes 3D ligand generation and binding affinity prediction. It integrates pocket-aware ligand design with multi-endpoint affinity prediction, offering a unified framework for structure-based drug design.
2. The model supports multiple design modes, including de novo generation, interaction/pharmacophore-conditional sampling, fragment elaboration, and multi-endpoint affinity prediction. This versatility makes it suitable for various stages of drug discovery, from hit identification to lead optimization.
3. Flowr.root achieves state-of-the-art performance in both unconditional 3D molecule and pocket-conditional ligand generation. It produces geometrically realistic, low-strain structures with high computational efficiency, outperforming recent models on established benchmark datasets.
4. The integrated affinity prediction module demonstrates superior accuracy on the SPINDR test set and outperforms recent models on the Schrödinger FEP /OpenFE benchmark, while offering substantial speed advantages.
5. As a foundation model, Flowr.root requires continuous parameter-efficient fine-tuning on project-specific datasets to account for unseen structure-activity landscapes. This approach yields strong correlation with experimental in-house data, making it a dynamic companion for early-stage drug discovery campaigns.
6. The model’s joint generation and affinity prediction capabilities enable inference-time scaling through importance sampling, effectively steering molecular design toward higher-affinity compounds. Case studies validate this approach, showing significant correlation between predicted and quantum-mechanical binding energies.
7. By integrating structure-aware generation, affinity estimation, and property-guided sampling within a unified framework, Flowr.root provides a comprehensive foundation for structure-based drug design, spanning hit identification through lead optimization.
📜Paper: arxiv.org/abs/2510.02578
#FlowrRoot #DrugDiscovery #StructureBasedDesign #LigandGeneration #AffinityPrediction
1
4
1,158
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
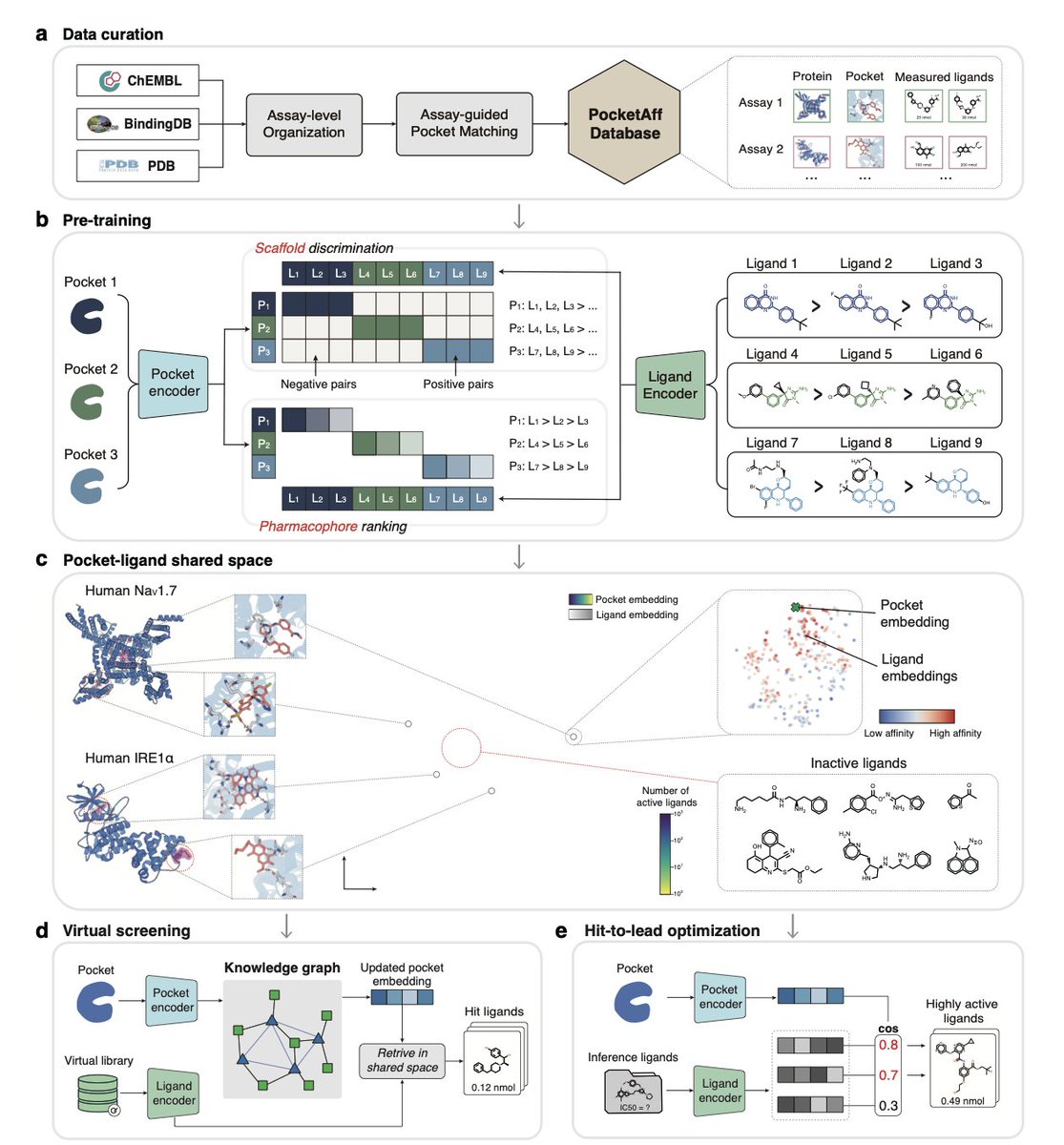
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
11
1,049
27 Jun 2025
Hierarchical affinity landscape navigation through learning a shared pocket-ligand space
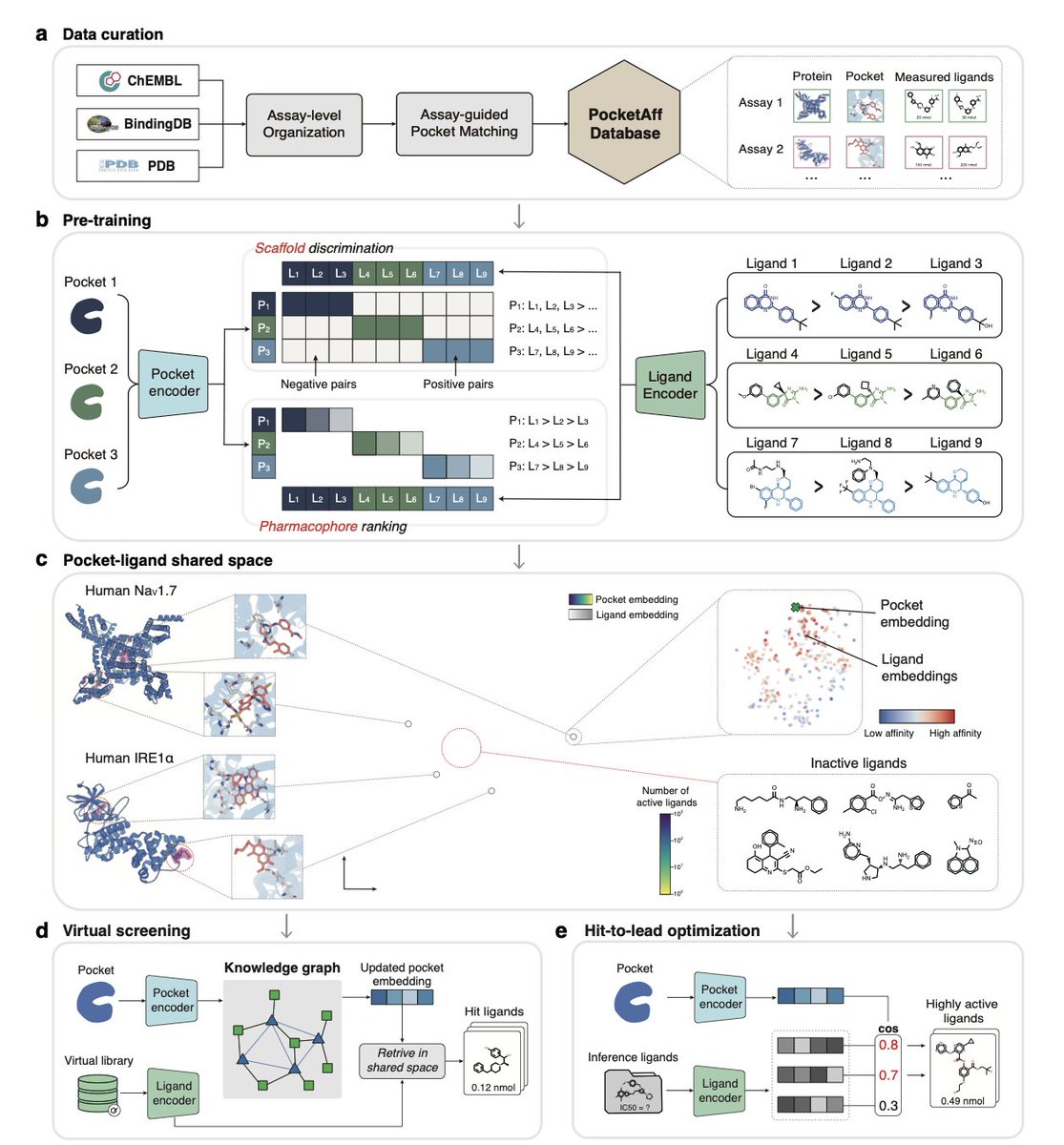
1.LigUnity is a foundation model for protein-ligand affinity prediction that unifies virtual screening and hit-to-lead optimization in a single shared embedding space, capturing both broad scaffold-level and fine-grained pharmacophore-level ligand interactions.
2.Unlike existing models that treat virtual screening and lead optimization separately, LigUnity jointly embeds ligands and protein pockets using scaffold discrimination and pharmacophore ranking to navigate a hierarchical affinity landscape.
3.In virtual screening, LigUnity outperforms 24 state-of-the-art methods across DUD-E, DEKOIS, and LIT-PCBA benchmarks, with over 50% improvement in EF1% and 10⁶× speedup compared to docking methods like Glide-SP, without requiring binding poses.
4.The model maintains high performance even on novel targets with low sequence similarity (<30%) to training proteins, demonstrating robust generalization capabilities that surpass both structure-based and structure-free baselines.
5.For hit-to-lead optimization, LigUnity outperforms physics-based methods such as FEP and structure-based models like GenScore on Merck and JACS FEP benchmarks, showing strong predictive power in zero-shot and few-shot scenarios.
6.Even under challenging conditions where both ligands and proteins are dissimilar to training data, LigUnity improves r² by 38.1% over its sequence-only variant, confirming the value of incorporating explicit pocket structure.
7.Fine-tuning LigUnity with only partial binding data (as few as 4–16 ligands) yields competitive or superior accuracy to commercial tools like FEP (OPLS4), offering an efficient alternative for large-scale lead optimization.
8.To support the model, the authors curated PocketAffDB, the largest structure-aware binding assay dataset, with 0.8M affinity datapoints, 0.5M unique ligands, and 53,406 binding pockets—enabling structure-aware learning across diverse assays.
9.LigUnity includes a heterogeneous GNN that leverages a large pocket-ligand knowledge graph (16M pocket-pocket edges and 0.83M pocket-ligand edges) to refine query embeddings, improving screening performance by sharing information across similar pockets.
10.When integrated into an active learning framework for TYK2 optimization, LigUnity successfully identifies high-affinity ligands within four iterations, achieving 40% r² improvement and discovering nanomolar hits with dramatically fewer FEP calculations.
11.The model is interpretable: through residue and atom-level masking, LigUnity highlights pharmacophoric groups and pocket residues crucial for binding, aligning well with known crystallographic interactions.
12.Across split-by-time, split-by-scaffold, and split-by-unit settings in ChEMBL and BindingDB, LigUnity consistently outperforms other models, particularly excelling in underexplored settings like percentage-based assay formats.
13.LigUnity eliminates the need for 3D docking or pose generation, making it a practical and fast solution for real-world drug discovery pipelines that involve millions of ligands and diverse protein targets.
14.The study presents LigUnity as a general-purpose, structure-aware foundation model for computer-aided drug discovery, bridging the gap between early virtual screening and downstream optimization with a single efficient architecture.
💻Code: github.com/IDEA-XL/LigUnity
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #MachineLearning #VirtualScreening #DeepLearning #Bioinformatics #StructureBasedDesign #LigUnity #ComputationalBiology #AffinityPrediction
4
32
1,810
18 Jun 2025
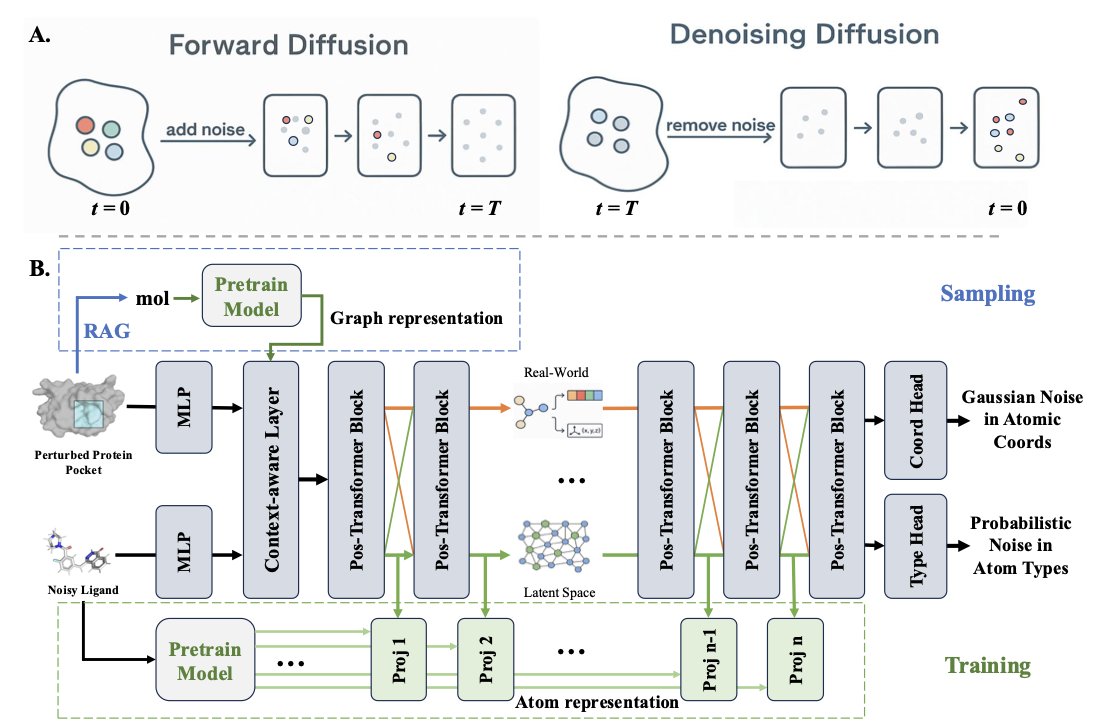
Reimagining Target-Aware Molecular Generation through Retrieval-Enhanced Aligned Diffusion
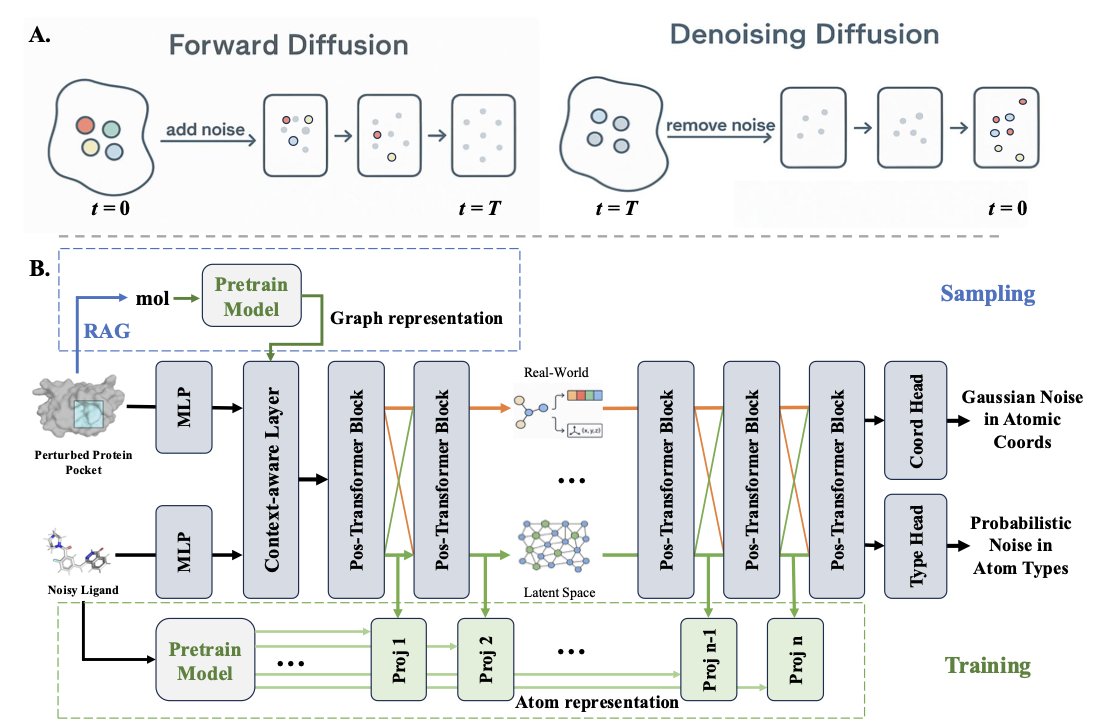
1.The paper introduces READ, the first framework that integrates Retrieval-Augmented Generation with an SE(3)-equivariant diffusion model for ligand design. It sets a new state of the art in structure-based molecule generation by aligning diffusion processes with chemical and geometric priors.
2.READ innovatively uses a contrastively pretrained encoder to learn a latent space embedding drug-like molecules, enabling atom-aware and geometry-aware guidance during generation. This eliminates the need for handcrafted chemical validity rules.
3.At inference time, READ retrieves ligands from a protein–ligand graph built using TM-align and DaliLite. Retrieved templates are selected based on pocket similarity and their embeddings guide the reverse diffusion process, ensuring pocket-specific, valid ligand synthesis.
4.This hierarchical retrieval strategy improves both exploration and exploitation. The model aligns retrieved molecular templates with diffusion steps via cross-modal attention, which enables READ to recover chemically realistic and geometrically compatible structures.
5.READ operates in a dual diffusion space—atomic coordinates and atom types—using stochastic differential equations. Retrieved embeddings influence both spaces to simultaneously enforce shape complementarity and valid atom typing.
6.Compared to 12 state-of-the-art baselines on the CBGBench benchmark, READ-2k achieves top performance in docking energy (Vina score), interaction fidelity (PLIP analysis), and chemical realism (low substructure divergence and steric clashes), outperforming native ligands in many cases.
7.An ablation study shows that retrieval alone adds little to unaligned models, and alignment alone helps modestly. Only when retrieval is combined with latent alignment does the model achieve significant improvements, confirming the synergy of the two components.
8.The generated ligands consistently outperform native ligands in docking energy across all evaluation protocols (Score, Minimize, Dock), and achieve improvement rates above 96% while preserving drug-likeness metrics such as QED, SA, and Lipinski rules.
9.READ achieves high sample validity (over 98%), fast convergence, and competitive sampling speed (~18–35 minutes for 100 ligands), with minimal overhead from the retrieval mechanism, which makes it practical for real-world drug design workflows.
10.Unlike voxel-based or autoregressive models, READ does not suffer from rotational asymmetry or steric clashes, and its chemically-informed latent space directly regularizes the denoising process, removing the need for post-generation filtering or optimization.
11.The method also shows promise for extensibility. Future work may scale the retrieval database with generated ligands, incorporate differentiable scoring into training, or account for receptor flexibility, indicating strong potential for generalization.
📜Paper: arxiv.org/abs/2506.14488
#DrugDiscovery #MolecularGeneration #DiffusionModels #ComputationalBiology #ProteinLigand #StructureBasedDesign
1
16
936
18 Jun 2025
Reimagining Target-Aware Molecular Generation through Retrieval-Enhanced Aligned Diffusion
1.The paper introduces READ, the first framework that integrates Retrieval-Augmented Generation with an SE(3)-equivariant diffusion model for ligand design. It sets a new state of the art in structure-based molecule generation by aligning diffusion processes with chemical and geometric priors.
2.READ innovatively uses a contrastively pretrained encoder to learn a latent space embedding drug-like molecules, enabling atom-aware and geometry-aware guidance during generation. This eliminates the need for handcrafted chemical validity rules.
3.At inference time, READ retrieves ligands from a protein–ligand graph built using TM-align and DaliLite. Retrieved templates are selected based on pocket similarity and their embeddings guide the reverse diffusion process, ensuring pocket-specific, valid ligand synthesis.
4.This hierarchical retrieval strategy improves both exploration and exploitation. The model aligns retrieved molecular templates with diffusion steps via cross-modal attention, which enables READ to recover chemically realistic and geometrically compatible structures.
5.READ operates in a dual diffusion space—atomic coordinates and atom types—using stochastic differential equations. Retrieved embeddings influence both spaces to simultaneously enforce shape complementarity and valid atom typing.
6.Compared to 12 state-of-the-art baselines on the CBGBench benchmark, READ-2k achieves top performance in docking energy (Vina score), interaction fidelity (PLIP analysis), and chemical realism (low substructure divergence and steric clashes), outperforming native ligands in many cases.
7.An ablation study shows that retrieval alone adds little to unaligned models, and alignment alone helps modestly. Only when retrieval is combined with latent alignment does the model achieve significant improvements, confirming the synergy of the two components.
8.The generated ligands consistently outperform native ligands in docking energy across all evaluation protocols (Score, Minimize, Dock), and achieve improvement rates above 96% while preserving drug-likeness metrics such as QED, SA, and Lipinski rules.
9.READ achieves high sample validity (over 98%), fast convergence, and competitive sampling speed (~18–35 minutes for 100 ligands), with minimal overhead from the retrieval mechanism, which makes it practical for real-world drug design workflows.
10.Unlike voxel-based or autoregressive models, READ does not suffer from rotational asymmetry or steric clashes, and its chemically-informed latent space directly regularizes the denoising process, removing the need for post-generation filtering or optimization.
11.The method also shows promise for extensibility. Future work may scale the retrieval database with generated ligands, incorporate differentiable scoring into training, or account for receptor flexibility, indicating strong potential for generalization.
📜Paper: arxiv.org/abs/2506.14488
#DrugDiscovery #MolecularGeneration #DiffusionModels #ComputationalBiology #ProteinLigand #StructureBasedDesign
8
618
17 Jun 2025
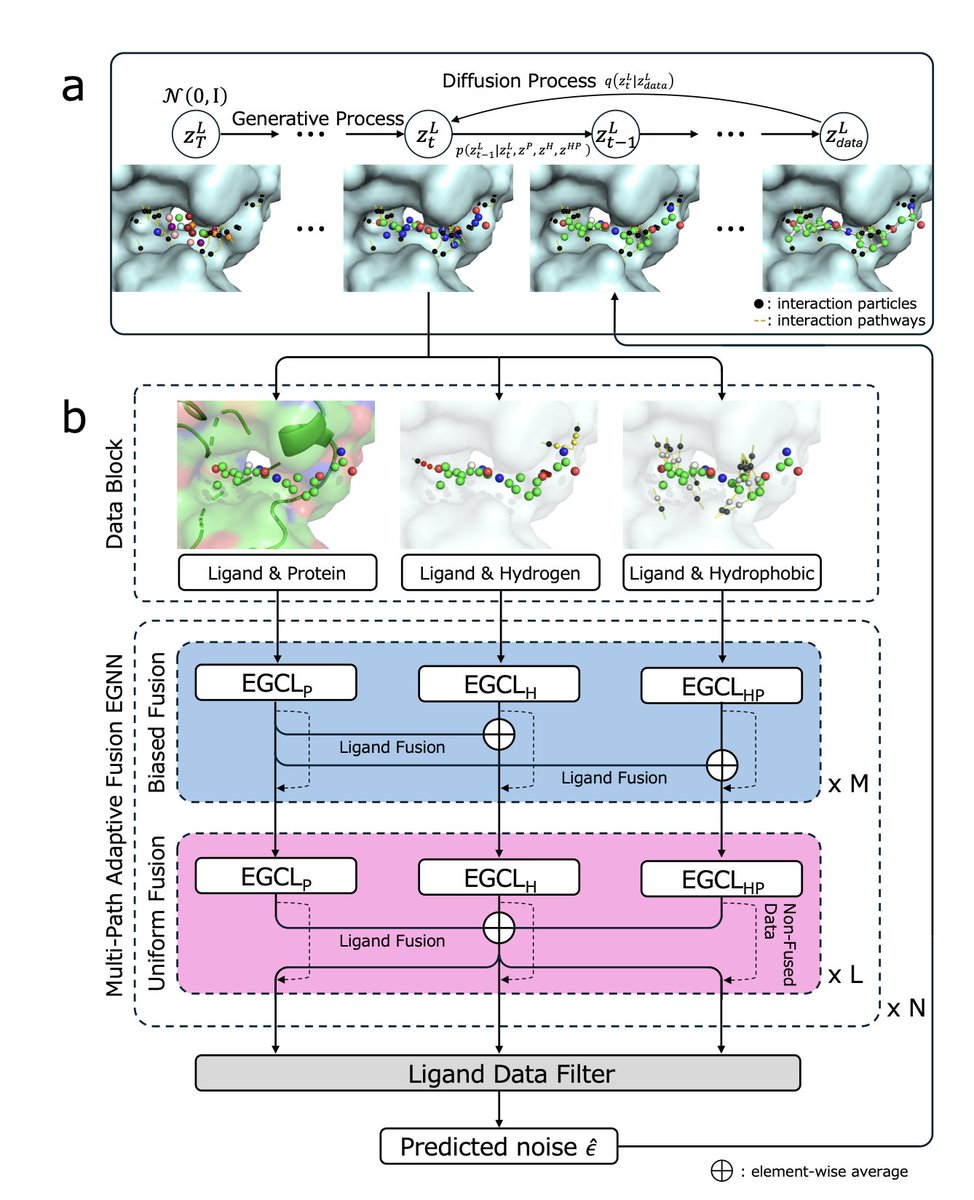
Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design
1.A novel 3D molecular generation framework, DiffPharma, is proposed to explicitly model key protein-ligand interactions—especially hydrogen bonds and hydrophobic contacts—using a conditional diffusion model guided by "interaction particles."
2.DiffPharma introduces "anchor" and "functional" interaction particles along predefined paths to accurately encode spatial and chemical constraints. These guide the generation process to ensure pharmacophore fidelity and spatial complementarity.
3.A multi-path adaptive fusion EGNN (MAP-EGNN) architecture integrates interaction-specific features via separate subnetworks for protein-ligand, hydrogen bond, and hydrophobic interactions. Two fusion mechanisms—biased and uniform—enable coherent information blending while retaining interaction-type specificity.
4.In benchmarks using the CrossDocked2020 dataset, DiffPharma achieves superior pharmacophore interaction reproducibility compared to Pocket2Mol, DeepICL, and DiffInt, with cosine similarity scores peaking near 0.9 for interaction pattern recovery.
5.Case studies on AKT1 and β-lactamase targets demonstrate that DiffPharma can generate synthetically accessible molecules that preserve key interaction motifs of reference ligands while displaying structural diversity and favorable QED scores.
6.A practical application to SARS-CoV-2 main protease (Mpro) shows that introducing or omitting specific interaction constraints (e.g., with GLU166) markedly affects the spatial distribution of interaction sites, pose stability (via MD simulations), and binding affinities (via MM/GBSA).
7.Molecules generated with explicit GLU166 constraints showed lower RMSD and more favorable ΔG_bind values than those without. Several even outperformed the reference ligand in binding free energy while maintaining drug-likeness and ADMET profiles.
8.DiffPharma successfully de novo reproduces a known bioactive ligand (PDB: 7GBL) based solely on protein structure and interaction constraints, without exposure to the compound during training—highlighting its capability for structure-based pharmacophore design.
9.ADMET profiling of generated molecules using ADMETlab 3.0 reveals high compliance with drug-likeness thresholds across key parameters including MW, TPSA, BBB penetration, and synthetic accessibility, confirming the practical relevance of the generated compounds.
10.DiffPharma is positioned as a promising framework for interaction-guided molecular generation in early-stage drug design, balancing interaction fidelity, chemical validity, and synthetic feasibility in a unified diffusion-based architecture.
💻Code: github.com/sekijima-lab/Diff… 📜Paper: doi.org/10.26434/chemrxiv-20…
#DrugDiscovery #MolecularDesign #DiffusionModels #Pharmacophore #AI4Science #StructureBasedDesign #ComputationalBiology #MachineLearning #ADMET #SARSCoV2 #EGNN
1
9
37
3,262
10 Jun 2025
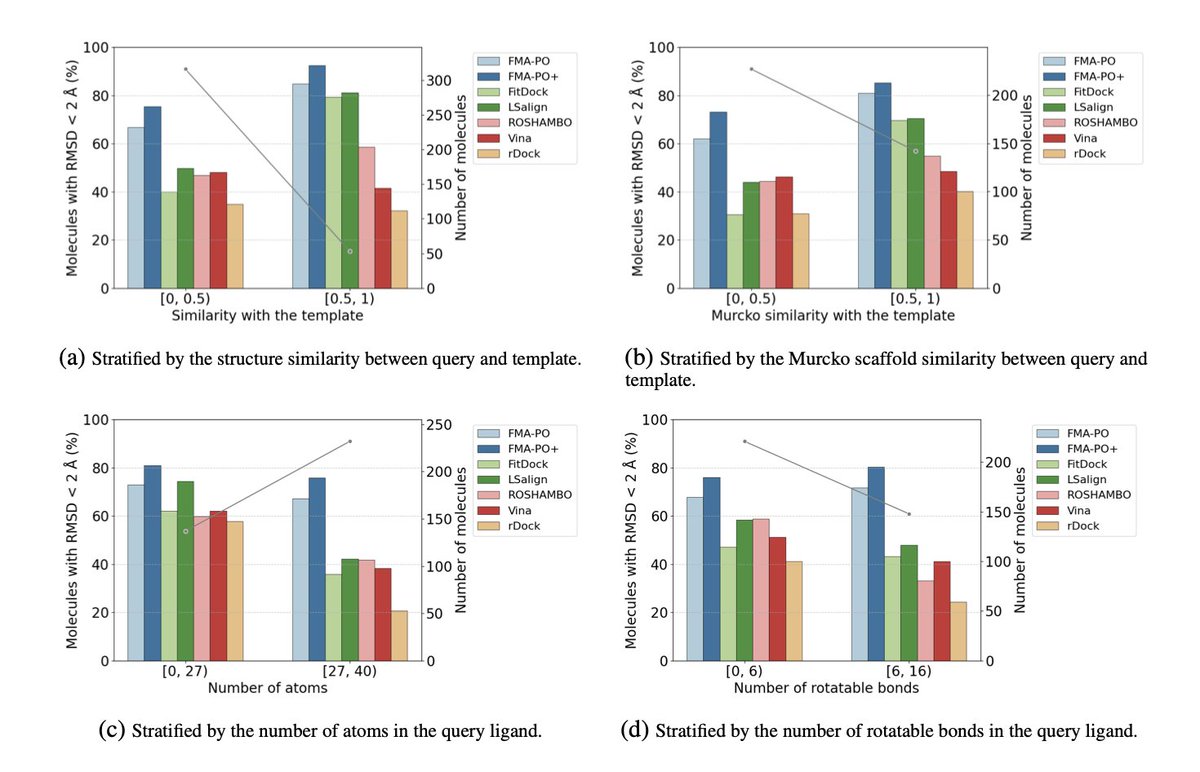
Template-Guided 3D Molecular Pose Generation via Flow Matching and Differentiable Optimization
1.The paper introduces FMA-PO, a novel two-stage method for 3D molecular pose prediction using a crystallized reference ligand as a template to guide ligand conformation generation from a 2D molecular graph.
2.The first stage applies a flow-matching generative model that progressively denoises random initial conformations to align query molecules spatially with the template ligand in 3D space.
3.The second stage refines initial poses by differentiable optimization directly on atomic coordinates, maximizing shape and pharmacophore similarity while minimizing internal energy and optionally considering protein pocket interactions.
4.FMA-PO uniquely integrates ligand-based alignment with receptor-aware refinement, leveraging both template ligand geometry and, optionally, protein binding site information to improve docking accuracy.
5.The method outperforms classical docking tools like AutoDock Vina and rDock, as well as recent open-access alignment methods, especially in challenging cases with low template-query similarity or highly flexible ligands.
6.The authors introduce AlignDockBench, a new benchmark dataset with 369 protein-ligand pairs spanning diverse targets, enabling realistic evaluation of template-based docking and alignment methods.
7.FMA-PO demonstrates higher accuracy with about 70% of predicted poses under 2Å RMSD, improving to nearly 78% with the enhanced FMA-PO version that refines all candidate poses.
8.The model architecture features a hybrid molecular graph including atomic and functional group nodes, processed by multi-head attention with edge bias and a vector field network predicting ligand conformations conditioned on the template.
9.Optimization uses novel shape-based and pharmacophore-based Tanimoto similarity scores computed via Gaussian volume overlaps, combined with internal energy from GAFF2 force fields, allowing flexible yet physically plausible pose adjustments.
10.The framework supports data augmentation and downstream predictive modeling, providing a pathway for improved structure-based drug design workflows that leverage known ligand geometries.
11.Future directions include incorporating receptor flexibility, conditioning on multiple templates, and enhancing computational efficiency to make the approach more practical for large-scale drug discovery.
💻Code: anonymous.4open.science/r/Al…
📜Paper: arxiv.org/abs/2506.06305v1
#DrugDesign #MolecularDocking #FlowMatching #StructureBasedDesign #ComputationalChemistry #DeepLearning #3DAlignment #LigandPosePrediction
1
6
736
10 Jun 2025
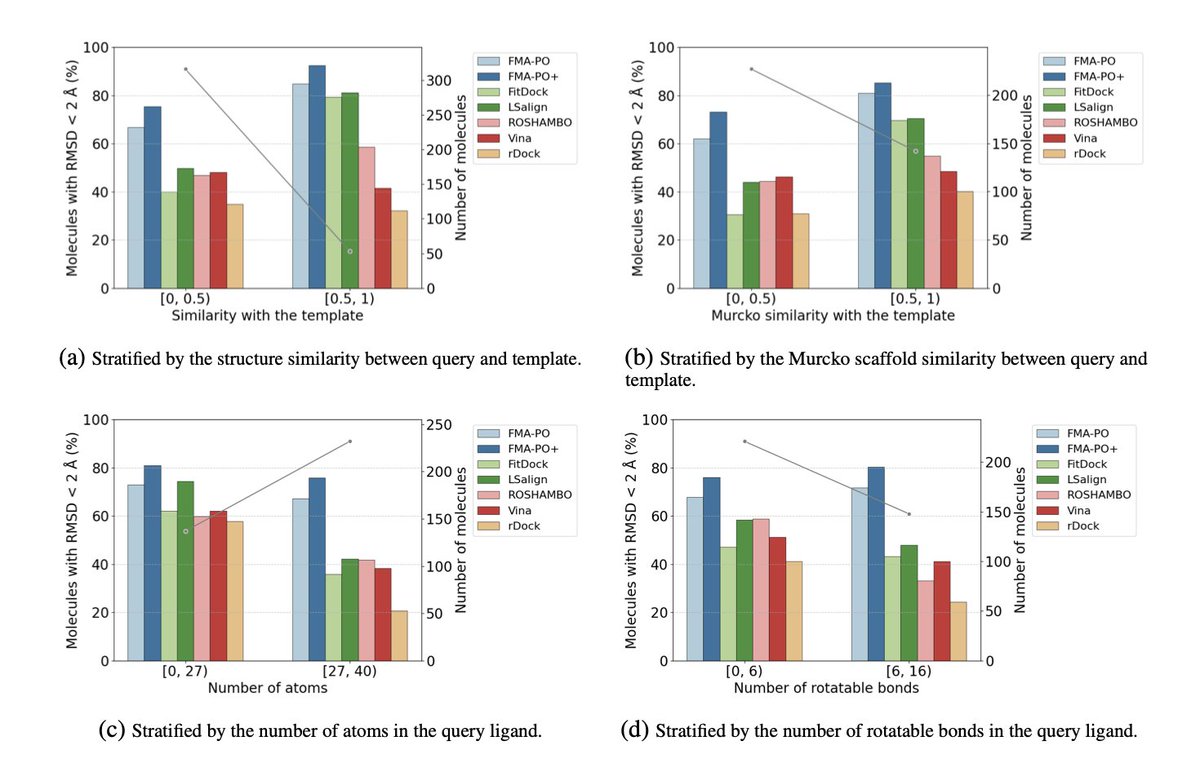
Template-Guided 3D Molecular Pose Generation via Flow Matching and Differentiable Optimization
1.The paper introduces FMA-PO, a novel two-stage method for 3D molecular pose prediction using a crystallized reference ligand as a template to guide ligand conformation generation from a 2D molecular graph.
2.The first stage applies a flow-matching generative model that progressively denoises random initial conformations to align query molecules spatially with the template ligand in 3D space.
3.The second stage refines initial poses by differentiable optimization directly on atomic coordinates, maximizing shape and pharmacophore similarity while minimizing internal energy and optionally considering protein pocket interactions.
4.FMA-PO uniquely integrates ligand-based alignment with receptor-aware refinement, leveraging both template ligand geometry and, optionally, protein binding site information to improve docking accuracy.
5.The method outperforms classical docking tools like AutoDock Vina and rDock, as well as recent open-access alignment methods, especially in challenging cases with low template-query similarity or highly flexible ligands.
6.The authors introduce AlignDockBench, a new benchmark dataset with 369 protein-ligand pairs spanning diverse targets, enabling realistic evaluation of template-based docking and alignment methods.
7.FMA-PO demonstrates higher accuracy with about 70% of predicted poses under 2Å RMSD, improving to nearly 78% with the enhanced FMA-PO version that refines all candidate poses.
8.The model architecture features a hybrid molecular graph including atomic and functional group nodes, processed by multi-head attention with edge bias and a vector field network predicting ligand conformations conditioned on the template.
9.Optimization uses novel shape-based and pharmacophore-based Tanimoto similarity scores computed via Gaussian volume overlaps, combined with internal energy from GAFF2 force fields, allowing flexible yet physically plausible pose adjustments.
10.The framework supports data augmentation and downstream predictive modeling, providing a pathway for improved structure-based drug design workflows that leverage known ligand geometries.
11.Future directions include incorporating receptor flexibility, conditioning on multiple templates, and enhancing computational efficiency to make the approach more practical for large-scale drug discovery.
💻Code: anonymous.4open.science/r/Al…
📜Paper: arxiv.org/abs/2506.06305v1
#DrugDesign #MolecularDocking #FlowMatching #StructureBasedDesign #ComputationalChemistry #DeepLearning #3DAlignment #LigandPosePrediction
2
5
745
27 May 2025
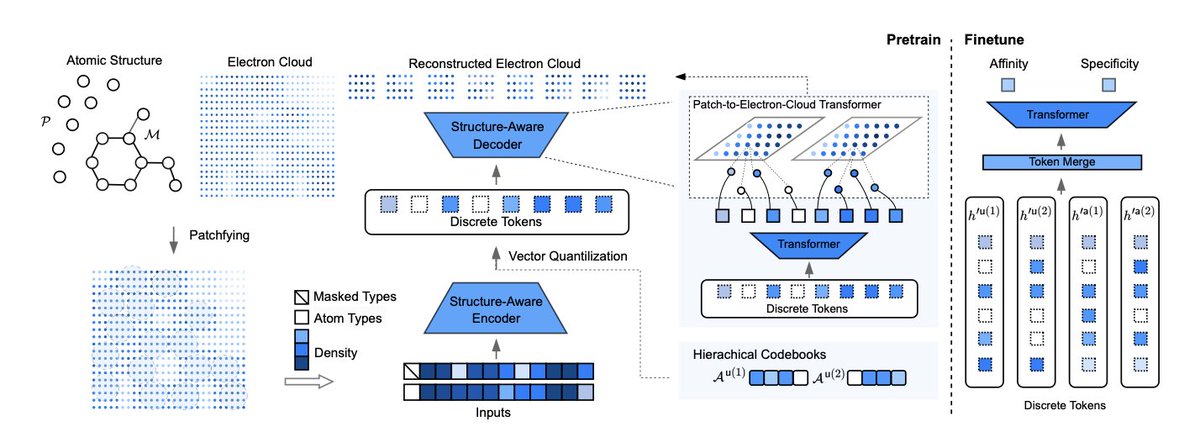
Tokenizing Electron Cloud in Protein-Ligand Interaction Learning
1.This paper presents ECBind, a novel framework that tokenizes electron cloud signals into quantized embeddings to enhance protein-ligand interaction prediction, uncovering binding patterns beyond atomic structure and achieving state-of-the-art results across multiple benchmarks.
2.Unlike conventional models that rely on atoms or fragments, ECBind explicitly incorporates electron density information—capturing non-covalent interactions, charge polarization, and π-π stacking—to provide a more physically grounded understanding of binding affinity and specificity.
3.A structure-aware transformer and hierarchical vector quantization are used to compress dense electron cloud data into discrete tokens, which are then used as inputs for downstream tasks such as binding affinity regression and activity classification.
4.ECBind is trained using a pretrain-finetune paradigm. During pretraining, the model reconstructs electron cloud signals and predicts masked atom types, enabling joint learning of 3D electronic and 2D atomic structures.
5.To address the high computational cost of computing electron clouds, the authors introduce ECBind-stdt, a student model trained via knowledge distillation from the full ECBind model, achieving nearly identical performance without requiring electron cloud inputs at inference.
6.On the MISATO dataset for relative binding affinity, ECBind-ptn achieves a 6.42% improvement in per-structure Pearson correlation and 15.58% in Spearman correlation over the best baseline, significantly outperforming fragment-level, atom-level, and hierarchical GNNs.
7.ECBind’s performance on LBA and LEP datasets also tops existing baselines. While the performance gain on LBA is more modest, this is attributed to LBA’s noisier electron cloud data, emphasizing the importance of data quality for quantum-scale modeling.
8.An ablation analysis shows that electron cloud tokenization is especially valuable for distinguishing subtle differences in ligand binding to the same protein, improving per-structure metrics by avoiding encoder over-smoothing.
9.By combining atomic and electronic structure representations, ECBind learns complementary features that enhance its capacity to model protein-ligand interactions holistically, especially in complex or "hard" binding sites.
10.The electron-aware encoder architecture is adapted from ESM3’s GeoMHA, enabling message passing that accounts for both geometric and invariant attention, and ensuring trans-rotational invariance in the learned embeddings.
11.Electron cloud patches are assigned to atoms via probabilistic sampling and neighborhood search, and are compressed into hierarchical codebooks for 2D atomic and 3D electronic attributes—enabling efficient, multi-scale representation learning.
12.ECBind sets a new direction in computational drug discovery by integrating quantum-level signals into deep learning pipelines without compromising scalability, enabling more accurate affinity predictions and ligand prioritization.
💻Code:
github.com/haitaoLin-git/ECB…
📜Paper:
arxiv.org/abs/2505.19014
#ProteinLigandBinding #QuantumML #ElectronDensity #ECBind #DrugDiscovery #AffinityPrediction #GraphNeuralNetworks #StructureBasedDesign #MolecularRepresentation #Bioinformatics
1
2
25
2,609
18 May 2025
MedSAGE: Bridging Generative AI and Medicinal Chemistry for Structure-Based Design of Small Molecule Drugs
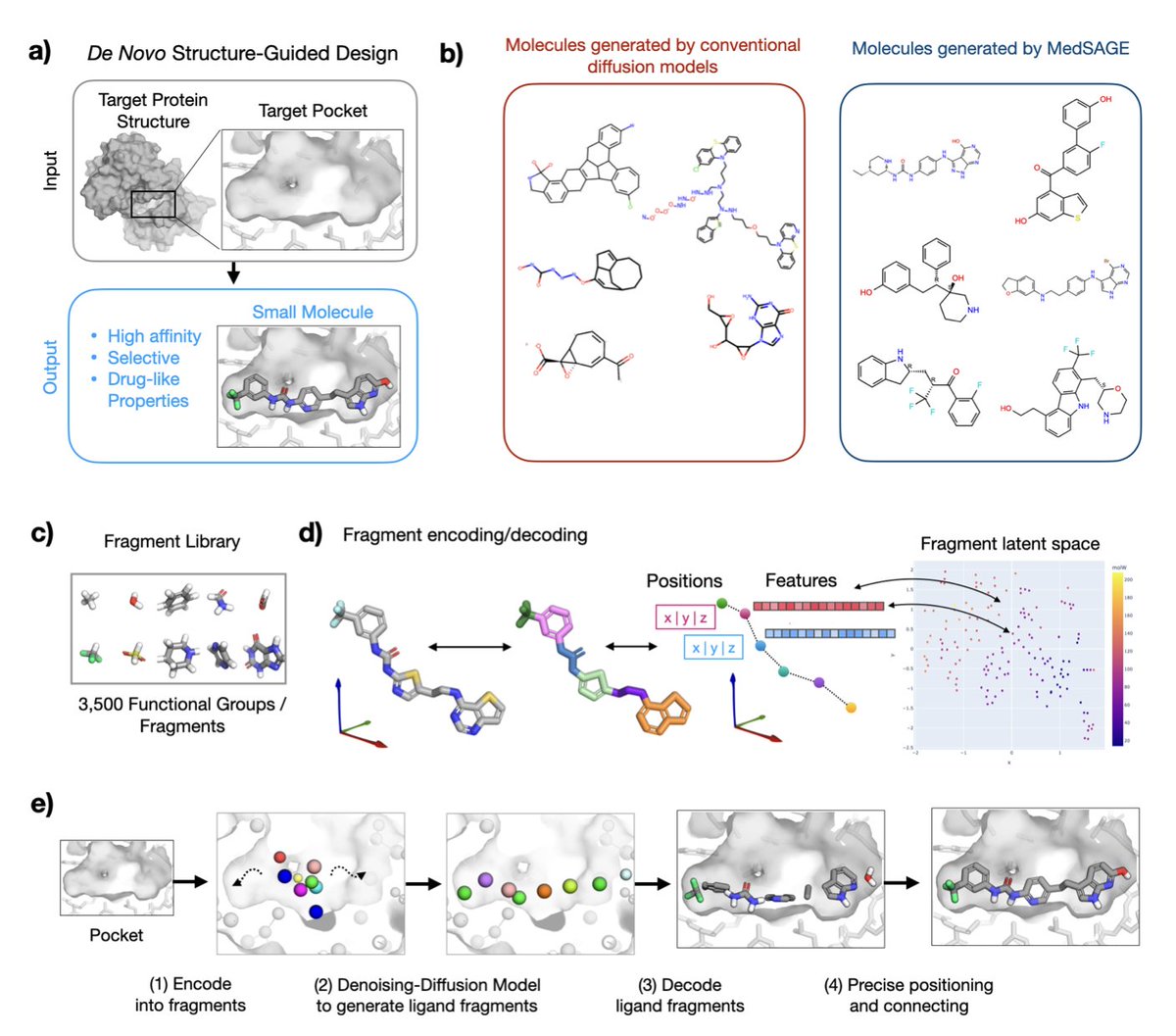
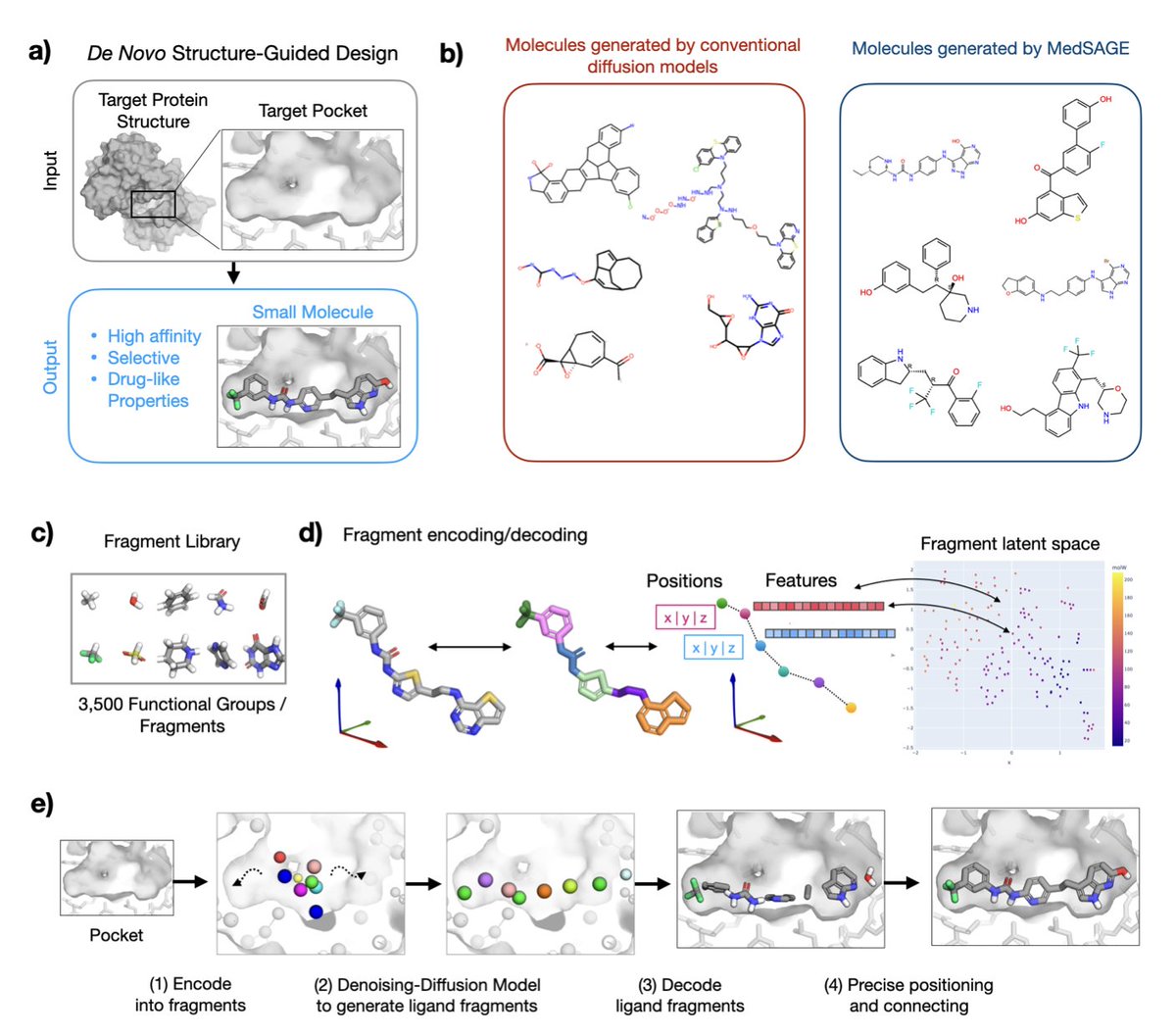
1.MedSAGE is a novel generative AI framework that addresses long-standing limitations in structure-based small-molecule drug design by operating directly on medicinal chemistry fragments instead of atoms or SMILES strings.
2.Unlike previous diffusion models that often generate chemically unstable or synthetically infeasible molecules, MedSAGE embeds functional groups and ring systems into a smooth latent space—improving interpretability, synthesizability, and design relevance.
3.The model employs a two-phase design: fragment generation via a 3D diffusion model guided by the protein pocket, followed by atomic-level assembly using a custom bond-connection algorithm that enforces chemical rules and optimizes Glide docking scores.
4.On a benchmark of 25 therapeutically relevant targets, MedSAGE-generated molecules had predicted affinities and selectivity statistically indistinguishable from those of approved drugs and clinical candidates—outperforming recent all-atom diffusion methods.
5.MedSAGE shows high selectivity: its ligands consistently reproduced more native binding interactions and avoided excessive hydrophobicity or bulkiness, unlike many molecules from DiffSBDD or IPDiff that lacked pocket specificity.
6.Despite no explicit optimization, MedSAGE molecules adhered to drug-likeness heuristics: Lipinski’s Rule of 5, appropriate logP, low synthetic complexity scores (~3.5), and scaffold diversity (~197 unique scaffolds out of 400 molecules per target).
7.Case studies showed MedSAGE could “rediscover” scaffolds structurally close to known binders like reboxetine, temsavir, and AK1, preserving key pharmacophores and even enhancing binding interactions via new hydrogen bonds.
8.Compared to traditional virtual screening of 30–400 million compound libraries, MedSAGE achieved comparable or better hit quality by generating only 2,000 molecules—offering a 100–1,000× improvement in hit enrichment efficiency.
9.Its fragment-based generation ensures aromatic ring planarity and reduces stereocenter complexity, avoiding common issues in atom-wise models. Fragment embeddings were learned via t-SNE and encode both 3D and chemical properties.
10.Although MedSAGE doesn’t yet handle protein flexibility or apo structures, it performs robustly on holo structure-based tasks—a realistic setting for many early-stage drug discovery projects.
11.The study introduces a scalable pipeline to pair AI-based molecule generation with similarity searches in large commercial libraries (e.g., Enamine REALSpace) to find purchasable analogs with similar binding profiles.
12.MedSAGE provides a compelling proof of concept that generative AI can capture medicinal chemistry principles and generate practical drug-like compounds directly from protein structures, with high interpretability and minimal data.
📜Paper: biorxiv.org/content/10.1101/…
#MedSAGE #DrugDesign #GenerativeAI #DiffusionModels #MedicinalChemistry #ProteinLigand #StructureBasedDesign #MolecularGeneration #FragmentBasedDesign #AI4Science #DeNovoDrugs #VirtualScreening #Bioinformatics #MachineLearning
1
18
76
5,721
18 May 2025
MedSAGE: Bridging Generative AI and Medicinal Chemistry for Structure-Based Design of Small Molecule Drugs
1.MedSAGE is a novel generative AI framework that addresses long-standing limitations in structure-based small-molecule drug design by operating directly on medicinal chemistry fragments instead of atoms or SMILES strings.
2.Unlike previous diffusion models that often generate chemically unstable or synthetically infeasible molecules, MedSAGE embeds functional groups and ring systems into a smooth latent space—improving interpretability, synthesizability, and design relevance.
3.The model employs a two-phase design: fragment generation via a 3D diffusion model guided by the protein pocket, followed by atomic-level assembly using a custom bond-connection algorithm that enforces chemical rules and optimizes Glide docking scores.
4.On a benchmark of 25 therapeutically relevant targets, MedSAGE-generated molecules had predicted affinities and selectivity statistically indistinguishable from those of approved drugs and clinical candidates—outperforming recent all-atom diffusion methods.
5.MedSAGE shows high selectivity: its ligands consistently reproduced more native binding interactions and avoided excessive hydrophobicity or bulkiness, unlike many molecules from DiffSBDD or IPDiff that lacked pocket specificity.
6.Despite no explicit optimization, MedSAGE molecules adhered to drug-likeness heuristics: Lipinski’s Rule of 5, appropriate logP, low synthetic complexity scores (~3.5), and scaffold diversity (~197 unique scaffolds out of 400 molecules per target).
7.Case studies showed MedSAGE could “rediscover” scaffolds structurally close to known binders like reboxetine, temsavir, and AK1, preserving key pharmacophores and even enhancing binding interactions via new hydrogen bonds.
8.Compared to traditional virtual screening of 30–400 million compound libraries, MedSAGE achieved comparable or better hit quality by generating only 2,000 molecules—offering a 100–1,000× improvement in hit enrichment efficiency.
9.Its fragment-based generation ensures aromatic ring planarity and reduces stereocenter complexity, avoiding common issues in atom-wise models. Fragment embeddings were learned via t-SNE and encode both 3D and chemical properties.
10.Although MedSAGE doesn’t yet handle protein flexibility or apo structures, it performs robustly on holo structure-based tasks—a realistic setting for many early-stage drug discovery projects.
11.The study introduces a scalable pipeline to pair AI-based molecule generation with similarity searches in large commercial libraries (e.g., Enamine REALSpace) to find purchasable analogs with similar binding profiles.
12.MedSAGE provides a compelling proof of concept that generative AI can capture medicinal chemistry principles and generate practical drug-like compounds directly from protein structures, with high interpretability and minimal data.
📜Paper: biorxiv.org/content/10.1101/…
#MedSAGE #DrugDesign #GenerativeAI #DiffusionModels #MedicinalChemistry #ProteinLigand #StructureBasedDesign #MolecularGeneration #FragmentBasedDesign #AI4Science #DeNovoDrugs #VirtualScreening #Bioinformatics #MachineLearning
1
4
868