Viral Proteins Reveal Geometry of Protein Language Models
1. The paper shows that protein language model (pLM) embedding spaces are dominated by a single “nativeness axis” (PC1) that strongly aligns with masked-reconstruction perplexity (a model-relative measure of how in-distribution a sequence is). This axis orders sequences from well-modeled cellular proteins to viral proteins to shuffled/random controls.
2. In ESMC-600M, PC1 explains 73.1% of embedding variance and correlates with perplexity at Spearman ρ=0.961, indicating that reconstruction difficulty is not just a score but a major geometric direction organizing embeddings across the tree of life.
3. Viral proteins are not treated as extreme outliers: they sit in an intermediate region—less “native” than cellular proteins but more structured than biologically meaningless sequences (position-shuffled or i.i.d. random), suggesting pLMs encode a continuum from in-distribution to out-of-distribution.
4. The same nativeness-axis geometry generalizes across ESM families (ESM2, ESMC, ESM3), including ESM3-OPEN (trained without viral sequences), and also appears in non-ESM architectures (ProGen2 autoregressive; EvoDiff discrete diffusion). This supports the idea that a dominant “model-fit” direction may be a broader property of sequence models trained on imbalanced biological data.
5. The work quantifies the underlying imbalance: UniRef50 pretraining coverage is heavily cellular-dominated (about 46.3M cellular clusters vs 390.3k viral; ~119× ratio), motivating the question of how underrepresented viral sequences are represented.
6. A key control argues nativeness is not simply “seen vs unseen”: cellular Swiss-Prot proteins released after an ESMC checkpoint (thus absent from its pretraining data) still look far more native-like (median PPL 5.3) than human viral proteins (median PPL 15.3), implying nativeness reflects compatibility with a cellular-dominated prior more than mere exposure.
7. Scaling changes viral nativeness, but unevenly across viral families: in ESMC (300M→6B), the fraction of human viral proteins below a native-like threshold (PPL<5) increases only modestly overall (~5%→~17%), while some families (e.g., Papillomaviridae, Retroviridae) shift strongly toward the native region and others (e.g., Orthomyxoviridae, Orthoherpesviridae, Sedoreoviridae) remain displaced.
8. Despite this dominant nativeness axis, embeddings retain viral-specific information beyond perplexity: linear probes on mean-pooled embeddings classify human viral vs cellular proteins with near-ceiling AUC (0.97–1.00 for larger models) under a homology-controlled split, and outperform both perplexity-only zero-shot classification and shallow sequence baselines (length, amino-acid composition, dipeptide composition).
9. The separation is especially relevant at low false-positive rates (screening-like settings): at 1% FPR, embedding probes achieve much higher TPR than perplexity-only classifiers (e.g., ESMC-6B 96.7% vs 39.2%), showing that “viral identity” is linearly accessible even when perplexity becomes a weaker separator at large scale.
10. Implications: nativeness (perplexity / PC1 position) can act as a diagnostic for where pLMs may be less reliable (notably for certain viral families), while embedding-based signals may complement homology methods for viral detection and biosecurity screening—though the authors emphasize evaluation and safety framing over deployment.
💻Code: github.com/MisteFr/viral-pro…
📜Paper: arxiv.org/abs/2606.12609
#ProteinLanguageModels #ESM #ViralProteins #RepresentationLearning #ComputationalBiology #Bioinformatics #MachineLearning #Biosecurity #Interpretability #ScalingLaws

4
47
3,582
hou zard retweeted

13 Aug 2024
Which protein binder design AI models will stand the test of time?
Here, we see:
EvoDiff ColabFold OpenMM HADDOCK for antibody discovery
biorxiv.org/content/10.1101/…
18
53
3,568
LineageFlow: Flow Matching for High-Fidelity Family-Aware Protein Sequence Generation
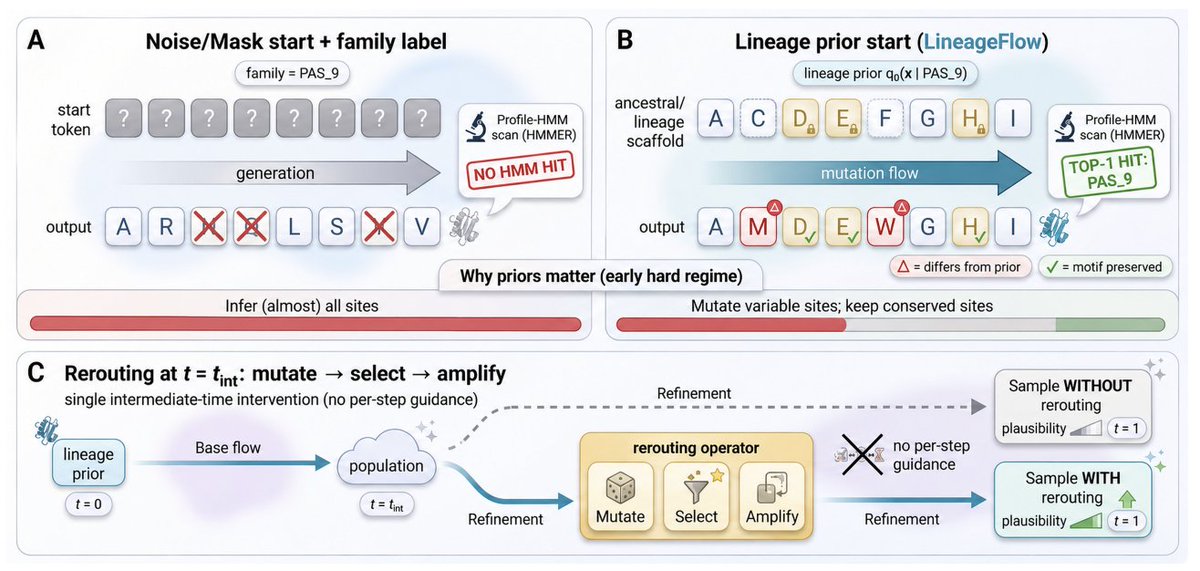
1. The paper argues that a key bottleneck in family-conditioned protein generation is the initialization prior: uniform-simplex noise or mask corruption erases evolutionary structure, forcing models to reconstruct conserved motifs “from scratch,” which weakens family control and plausibility.
2. LineageFlow replaces generic priors with lineage priors derived from ancestral sequence reconstruction (ASR): for each Pfam family, it infers a phylogeny from the MSA, performs marginal ASR at the root, and converts the site-wise root posterior into Dirichlet parameters used as a family-specific prior over the probability simplex.
3. With this design, generation is reframed as structured mutation from an evolved scaffold: conserved positions start concentrated, while variable sites retain uncertainty, aligning the trajectory with a family-specific manifold without feeding family labels or MSA prompts into the denoiser.
4. Methodologically, it builds on Dirichlet Flow Matching (DFM) on the simplex: each site follows an analytic Dirichlet path Dir(α(h,l) (tmax t) ei), with a derived lineage-specific vector field that conserves probability mass and keeps trajectories on the simplex.
5. Training uses a classifier parameterization: a transformer denoiser (initialized from ESM2) predicts terminal residues given (Xt, t), optimized by cross-entropy on valid (non-gap) MSA positions; the drift field is reconstructed by mixing analytic per-residue fields weighted by the predicted terminal distribution.
6. A second contribution is rerouting: a single intermediate-time inference intervention inspired by directed evolution (mutate → select → amplify) that steers samples toward a fitness objective without per-step gradient guidance, formalized as KL-regularized exponential tilting of the intermediate distribution.
7. Large-scale evaluation trains one shared model across 8,886 Pfam families (~8.94M sequences; 5% held-out per family) and scores generation by profile-HMM family validity (HMMER), foldability proxy (OmegaFold pLDDT), self-consistency (ESM-IF perplexity), novelty (MMseqs2 NN identity), and diversity (MMseqs2 clustering).
8. Results emphasize the role of priors: uniform-/mask-initialized baselines (DFM, EvoDiff) show essentially zero Pfam top-1 family accuracy under this strict HMM library scan, even when given explicit family labels; ASR prior alone (iid sampling) already yields high family validity, indicating ASR carries strong family signal.
9. LineageFlow with rerouting achieves near-natural family validity (Accfam 95.3% vs 96.6% for held-out natural sequences), improves foldability over prior-only and over several baselines (mean pLDDT 76.6), while keeping substantial novelty among foldable samples (Novelty@0.8 86.2%, Novelty@0.6 48.9%) and strong diversity.
10. A mechanistic analysis attributes gains to the “hard regime” at early times: Bayes-oracle denoising accuracy is higher under ASR priors than uniform priors when states are most corrupted, raising the recoverable signal ceiling and reducing early errors that propagate through the flow.
11. In a zero-shot enzyme case study, the denoiser is trained without three enzyme families, but priors are still built from their MSAs/trees; sampling without fine-tuning preserves motifs and novelty, and rerouting (using an unsupervised ESM2 plausibility objective) increases motif agreement and improves solubility/thermostability proxy distributions.
12. Limitations noted: reliance on high-quality MSAs and phylogenetic inference for priors; generation is tied to family alignment coordinates and does not model indels explicitly; evaluation relies on computational proxies (pLDDT, predictor-based properties) without experimental validation; rerouting adds compute and depends on the fitness function.
💻Code: github.com/Jinx-byebye/Linea…
📜Paper: arxiv.org/abs/2605.22252
#ComputationalBiology #ProteinDesign #GenerativeModels #FlowMatching #DiffusionModels #Phylogenetics #AncestralSequenceReconstruction #MachineLearning #Bioinformatics
7
32
2,857
MP2D: Constrained Monte Carlo Tree-Guided Diffusion for Multi-Objective Protein Sequence Design
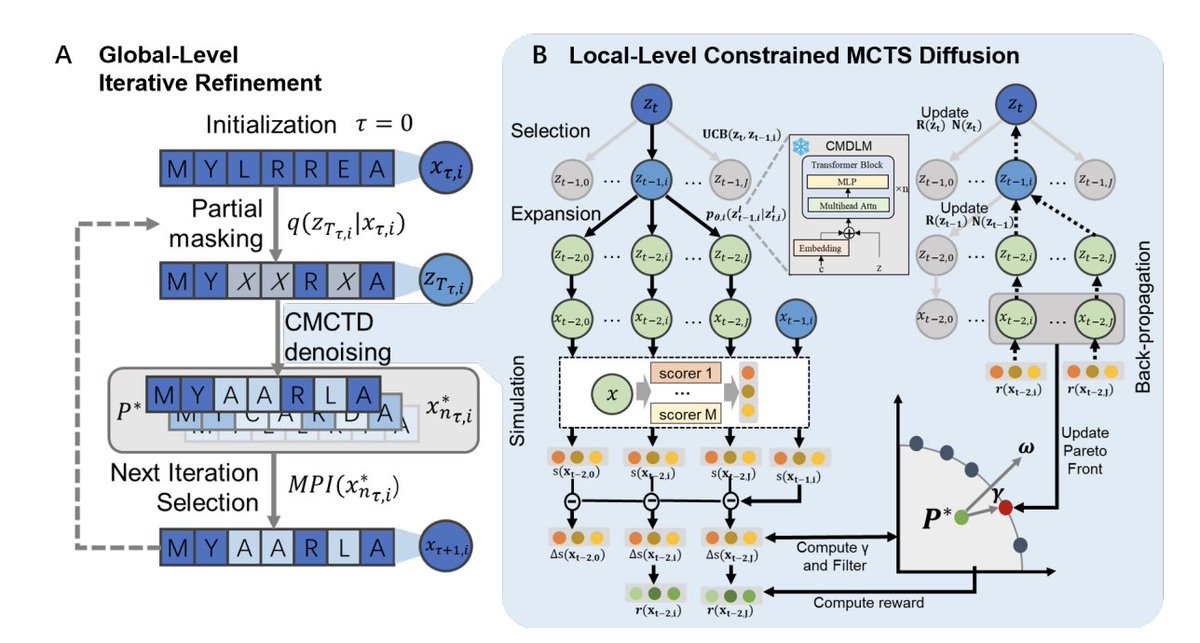
1 MP2D frames diffusion denoising as a constrained sequential decision-making problem and uses Monte Carlo Tree Search (MCTS) to explore many alternative denoising trajectories, rather than committing to a single diffusion path when objectives conflict (e.g., efficacy vs toxicity).
2 The key multi-objective ingredient is Pareto-guided search: each candidate is evaluated by a vector of property predictors, and MCTS selection/expansion is restricted to Pareto non-dominated children, so the search explicitly targets balanced trade-offs instead of relying on fragile scalar weights.
3 To keep Pareto optimization scalable when optimizing 4–5 objectives, MP2D introduces a dynamic Pareto constraint: candidate updates are filtered by alignment to predefined optimization directions (Das–Dennis simplex lattice) using an angular threshold that is adaptively tuned to maintain a stable rejection/acceptance rate, preventing Pareto-front “bloat” and property collapse.
4 MP2D is training-free at optimization time: it plugs in pretrained property evaluators and a pretrained conditional discrete diffusion model, and can swap objectives/predictors without retraining the generative backbone—positioned as a practical workflow for rapidly changing design specifications.
5 The generative backbone is CMDLM, a classifier-free, label-guided conditional masked diffusion language model for peptides (built on an ESM-style transformer). It is pretrained on 2.6M UniProt peptides (length 2–50) and then LoRA fine-tuned for antimicrobial peptides (AMPs) and protein binders (PBs), aiming to narrow the search space to task-relevant sequence regions.
6 CMDLM is evaluated on plausibility and realism using ESM-2 pseudoperplexity, structural foldability via OmegaFold pLDDT, and distributional similarity via Frechet ProtT5 Distance (FPD). Across peptide, AMP, and PB settings, CMDLM is generally more plausible/foldable and closer to target distributions than ProteinGAN, ProtGPT2, and EvoDiff under the paper’s benchmark.
7 The optimization engine (CMCTD) modifies UCB in MCTS by adding a diffusion-posterior–guided exploration term, encouraging exploration that stays close to valid diffusion transitions while still pursuing multi-property reward improvements.
8 MP2D adds a global iterative refinement loop: starting from W seed sequences, it repeatedly partially remasks sequences at random noise levels and reruns constrained MCTS denoising. This is designed to (a) correct early irreversible token decisions typical of single-pass denoising, and (b) increase diversity of optimization routes under noisy global property predictors.
9 On protein binder optimization (5 objectives: hemolysis, non-fouling, solubility, half-life, affinity) for targets 1B8Q and PPP5, MP2D outperforms classical multi-objective optimizers (NSGA-III, SMS-EMOA, SPEA2, MOPSO) and a recent generative baseline (MOG-DFM), achieving lower hemolysis, higher non-fouling/solubility/half-life, and competitive affinity.
10 On AMP optimization (4 objectives: antimicrobial probability, MIC, hemolysis, toxicity), MP2D outperforms AMP-focused multi-objective generative baselines (Multi-CGAN, MPOGAN, HMAMP, MoFormer), improving potency (higher Pamp, lower MIC) while simultaneously reducing safety risks (lower hemolysis and toxicity), addressing the common failure mode where optimizing one property degrades another.
📜Paper: arxiv.org/abs/2605.05829
#ProteinDesign #ComputationalBiology #DiffusionModels #MonteCarloTreeSearch #MultiObjectiveOptimization #AntimicrobialPeptides #PeptideDesign #ProteinEngineering #GenerativeAI #ParetoOptimization
2
27
1,865
EvoFlows: Evolutionary edit-based flow-matching for protein engineering
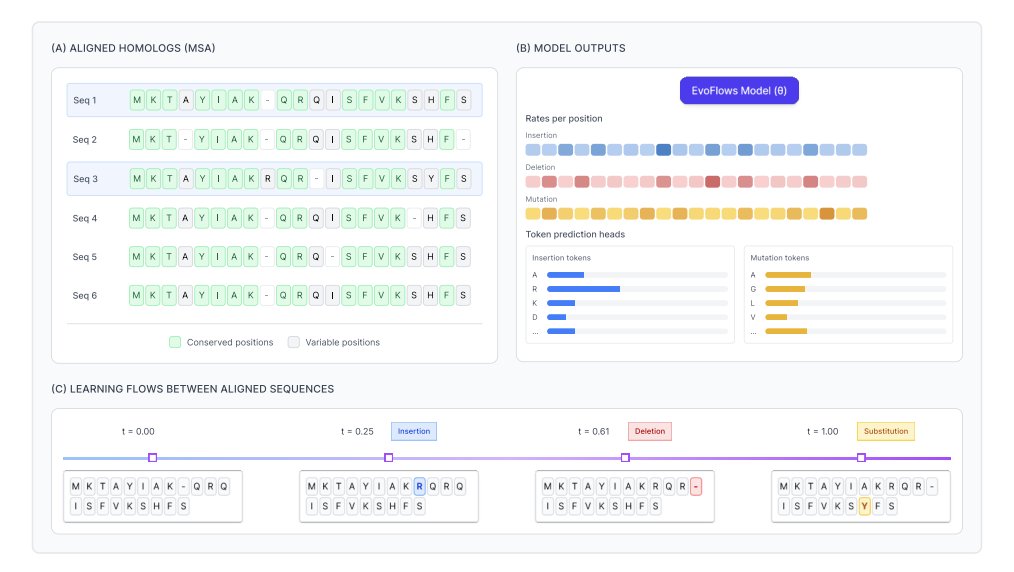
1 EvoFlows is a variable-length sequence-to-sequence generative model for protein optimization that edits an existing template via explicit elementary operations (substitutions, insertions, deletions), predicting both what mutation to apply and where—without pre-specifying mutation sites.
2 The paper argues current protein language model paradigms are misaligned with template-based optimization: autoregressive models require full regeneration and offer weak control over mutation count; masked LMs require externally chosen positions and do not natively support indels; discrete diffusion inherits similar conditioning/masking issues when starting from a template.
3 EvoFlows is built on discrete flow matching with edit flows (a CTMC over sequences) where transitions correspond to edit operations. This makes the generation path resemble realistic evolutionary/engineering trajectories: variants are reached through a controllable number of small, local edits rather than token-by-token regeneration or denoising.
4 Training data is constructed from natural homologs: for each seed protein, homolog sets are retrieved from UniRef30 (plus added evolutionary depth via an environmental database), then training pairs are formed by enumerating unordered pairs within each homolog set. Pairs are globally aligned with Needleman–Wunsch to introduce an explicit gap/empty token representation that enables modeling insertions and deletions.
5 A key methodological detail is inference: instead of grid-based Euler-style stepping with repeated Bernoulli sampling, EvoFlows samples event times directly from the model’s total rate via inverse-CDF with numerical integration, then samples the specific next edit from normalized rates. A length-normalized “clock normalization” hyperparameter provides control over expected edit counts independent of sequence length.
6 Architecture: EvoFlows uses the encoder trunk of a pretrained ESM-2 model to embed the current sequence, applies time conditioning late via FiLM (making time updates cheap), and uses shallow MLP heads to parameterize per-position rates and token distributions for substitution/insertion/deletion operations. ESM-2 weights are finetuned jointly rather than frozen.
7 Before natural-protein experiments, the paper introduces a deterministic synthetic benchmark with known ground-truth edit rules, enabling precise evaluation of whether the model recovers edit type, position, and identity. Results show reliable separation among no-op/insertion/substitution/deletion, and stable performance across sequence lengths.
8 In silico evaluation spans six diverse families (enzymes, growth factors, antibody fragments including VHH and scFv; datasets include Anti-SARS-CoV-2 VHH, Anti-EphA2 VHH, SPAT, FGF2, DhaA, Anti-HER2 scFv). Compared against EvoDiff-MSA, evotuned ESM-2 (with/without forced substitutions), random homolog pairing, and random mutations, EvoFlows matches holdout family distributions while exploring farther from the template and producing more diverse variant sets.
9 The analysis highlights practical indel handling: EvoFlows treats indels as first-class operations, while diffusion/MSA masking approaches can struggle to introduce insertions reliably (gap runs often regenerate as gaps; masking strategies complicate control of insertion count). MLM baselines are effectively limited to substitutions, making EvoFlows particularly relevant for indel-heavy families like antibodies.
📜Paper: arxiv.org/abs/2603.11703
#ProteinEngineering #GenerativeModels #ProteinLanguageModels #DiffusionModels #FlowMatching #Antibodies #ComputationalBiology #MachineLearning #ICLR2026
6
32
2,342
CMADiff: Cross-Modal Aligned Diffusion for Controllable Protein Generation
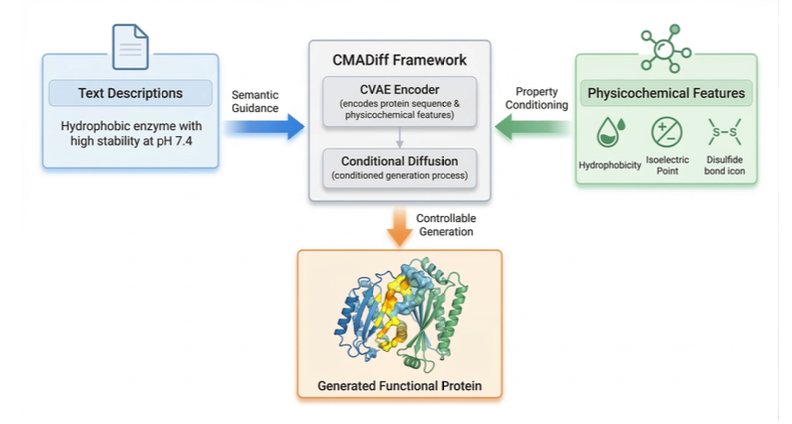
1. CMADiff introduces a cross‑modal framework that couples protein physicochemical features with natural‑language descriptors through a latent diffusion process, allowing users to steer sequence generation with textual prompts.
2. It is the first generative protein model to jointly embed physicochemical properties—such as hydrophobicity, charge, and disulfide propensity—and semantic context, leveraging a Conditional Variational Auto‑Encoder (CVAE) to encode both local and global biochemical signals into a rich latent space.
3. The BioAligner module aligns sentence embeddings (via Sentence‑BERT) with global physicochemical vectors using contrastive learning, providing a smooth, interpretable mapping that lets text descriptions directly influence the latent diffusion dynamics.
4. Conditional diffusion in the latent space, guided by BioAligner, yields sequences that match target physicochemical distributions, achieve AlphaFold‑3 pLDDT scores above 70 on average, and maintain TM‑scores >0.5, demonstrating high structural plausibility.
5. Empirical results show CMADiff surpasses state‑of‑the‑art baselines such as EvoDiff and TaxDiff on multiple metrics, achieving a novelty ratio of 0.78, a 13‑minute generation time for 1,000 sequences, and superior alignment between generated and natural physicochemical profiles.
6. Ablation studies confirm the essential roles of both the diffusion process and BioAligner: removing either component drops pLDDT by more than 10 %, underscoring their synergistic contribution to quality and controllability.
7. Future work plans to integrate explicit 3D geometric constraints into the diffusion pipeline, expand the model with multi‑omics data, and validate generated proteins experimentally, moving toward a fully interactive text‑to‑protein design platform.
💻Code: github.com/HPC-NEAU/PhysChem…
📜Paper: arxiv.org/abs/2503.21450
#ProteinDesign #DiffusionModels #Bioinformatics #AI #ProteinEngineering
1
3
15
1,585
Training‑free generation of protein sequences from small family alignments via stochastic attention
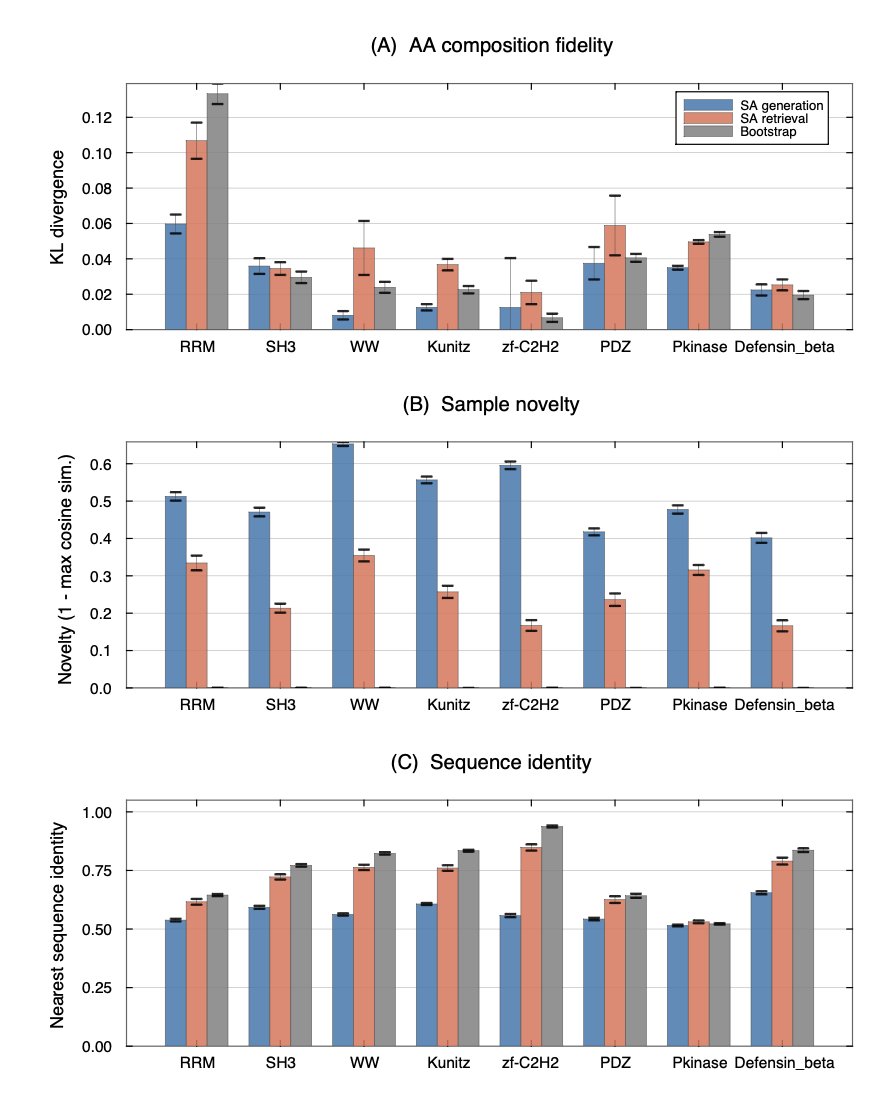
1 The authors introduce a training‑free sampler that treats a small protein family alignment as a modern Hopfield memory. A single soft‑max attention operation yields the exact energy gradient, so Langevin dynamics can explore the Boltzmann distribution without any learned parameters, GPU or back‑propagation.
2 Across eight Pfam families (37–420 members, 23–262 residues) the method produces sequences with amino‑acid KL divergence below 0.06, novelty 0.40–0.65, and predicted folds from ESMFold and AlphaFold2 that match the family templates (TM‑score improvements in 6 of 8 families, pLDDT up to 95).
3 The critical inverse temperature for the generation‑retrieval transition is predicted from the PCA dimensionality of the alignment: β* ≈ 1.57 0.28 √d, explaining 97 % of the variance. This simple linear rule lets the sampler run automatically with no temperature sweep.
4 Compared to profile HMMs, EvoDiff, and the MSA Transformer, stochastic attention keeps 51–66 % sequence identity to stored members while remaining highly novel, and it runs in seconds on a laptop versus hours of GPU time for pretrained baselines.
5 Independent checks show that the generated sequences preserve pairwise mutual information, satisfy a cationic, cysteine‑rich filter for β‑defensins, overlap 90 % with experimentally tolerated deep‑mutational‑scanning substitutions, and are scored as protein‑like by the 650‑million‑parameter ESM‑2 language model.
6 Limitations include fixed alignment length (no indels), dependence on a 95 % variance PCA cut‑off, and best performance on compact, well‑aligned domains; extending to variable‑length or highly divergent families remains future work.
7 The work demonstrates that the statistical structure of a small family is enough to generate realistic, novel proteins without any training, opening a low‑resource path for design in orphan or metagenomic families and suggesting broader applications to other scarce‑data domains.
💻Code: github.com/varnerlab/SA-Prot…
📜Paper: arxiv.org/abs/2603.14717
#ProteinDesign #DeepLearning #HopfieldNetworks #ProteinEngineering #ComputationalBiology
2
11
1,224
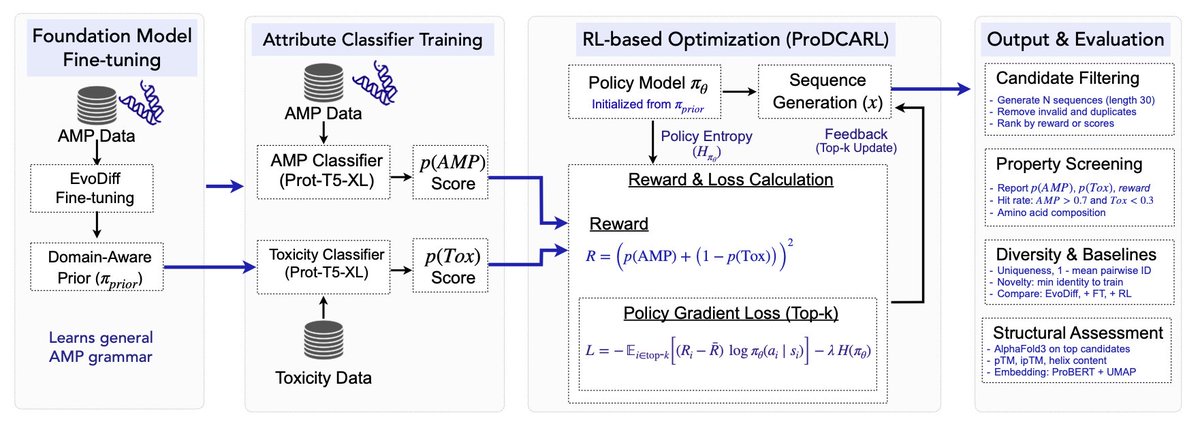
ProDCARL: Reinforcement Learning-Aligned Diffusion Models for De Novo Antimicrobial Peptide Design
1 ProDCARL couples EvoDiff OA-DM 38M with RL to directly optimize predicted antimicrobial activity while penalizing toxicity, pushing mean pAMP from 0.081 to 0.178 and lifting the joint high-quality hit rate to 6.3 %.
2 A top-k policy-gradient update focuses learning on the best 30 % of each batch; entropy regularization plus early-stopping at the first sign of diversity collapse keeps 1-mean pairwise identity at 0.929, avoiding mode collapse.
3 The pipeline first fine-tunes the diffusion prior on ~95 k AMP positives, then trains two ProtT5-XL-based classifiers (ROC-AUC 0.979 for AMP, 0.950 for toxicity) to supply the reward signal R = (pAMP 1 − pTox)².
4 Generated 30-aa peptides pass AlphaFold3 scrutiny: top hits adopt α-helical membrane-active folds typical of natural AMPs and cluster within the AMP-dense region of ProtBERT embedding space.
5 Fixed length, predictor-only rewards and limited toxicity data are acknowledged constraints; authors call for wet-lab assays, ensemble rewards and multi-objective guards before any therapeutic use.
💻Code: github.com/HIVE-UofT/ProDCAR…
📜Paper: arxiv.org/abs/2602.00157
#AI4Antimicrobials #DiffusionModels #ReinforcementLearning #PeptideDesign #AMR
1
4
18
1,685

4 Oct 2025
Microsoft says AI can create “zero day” threats in biology | Antonio Regalado, MIT Technology Review
Artificial intelligence can design toxins that evade security controls.
A team at Microsoft says it used artificial intelligence to discover a "zero day" vulnerability in the biosecurity systems used to prevent the misuse of DNA.
These screening systems are designed to stop people from purchasing genetic sequences that could be used to create deadly toxins or pathogens. But now researchers led by Microsoft’s chief scientist, Eric Horvitz, say they have figured out how to bypass the protections in a way previously unknown to defenders.
The team described its work today in the journal Science.
Horvitz and his team focused on generative AI algorithms that propose new protein shapes. These types of programs are already fueling the hunt for new drugs at well-funded startups like Generate Biomedicines and Isomorphic Labs, a spinout of Google.
The problem is that such systems are potentially “dual use.” They can use their training sets to generate both beneficial molecules and harmful ones.
Microsoft says it began a “red-teaming” test of AI’s dual-use potential in 2023 in order to determine whether “adversarial AI protein design” could help bioterrorists manufacture harmful proteins.
The safeguard that Microsoft attacked is what’s known as biosecurity screening software. To manufacture a protein, researchers typically need to order a corresponding DNA sequence from a commercial vendor, which they can then install in a cell. Those vendors use screening software to compare incoming orders with known toxins or pathogens. A close match will set off an alert.
To design its attack, Microsoft used several generative protein models (including its own, called EvoDiff) to redesign toxins—changing their structure in a way that let them slip past screening software but was predicted to keep their deadly function intact.
The researchers say the exercise was entirely digital and they never produced any toxic proteins. That was to avoid any perception that the company was developing bioweapons.
Before publishing the results, Microsoft says, it alerted the US government and software makers, who’ve already patched their systems, although some AI-designed molecules can still escape detection.
“The patch is incomplete, and the state of the art is changing. But this isn’t a one-and-done thing. It’s the start of even more testing,” says Adam Clore, director of technology R&D at Integrated DNA Technologies, a large manufacturer of DNA, who is a coauthor on the Microsoft report. “We’re in something of an arms race.”
To make sure nobody misuses the research, the researchers say, they’re not disclosing some of their code and didn’t reveal what toxic proteins they asked the AI to redesign. However, some dangerous proteins are well known, like ricin—a poison found in castor beans—and the infectious prions that are the cause of mad-cow disease.
“This finding, combined with rapid advances in AI-enabled biological modeling, demonstrates the clear and urgent need for enhanced nucleic acid synthesis screening procedures coupled with a reliable enforcement and verification mechanism,” says Dean Ball, a fellow at the Foundation for American Innovation, a think tank in San Francisco.
Ball notes that the US government already considers screening of DNA orders a key line of security. Last May, in an executive order on biological research safety, President Trump called for an overall revamp of that system, although so far the White House hasn’t released new recommendations.
Others doubt that commercial DNA synthesis is the best point of defense against bad actors. Michael Cohen, an AI-safety researcher at the University of California, Berkeley, believes there will always be ways to disguise sequences and that Microsoft could have made its test harder.
“The challenge appears weak, and their patched tools fail a lot,” says Cohen. “There seems to be an unwillingness to admit that sometime soon, we’re going to have to retreat from this supposed choke point, so we should start looking around for ground that we can actually hold.”
Cohen says biosecurity should probably be built into the AI systems themselves—either directly or via controls over what information they give.
But Clore says monitoring gene synthesis is still a practical approach to detecting biothreats, since the manufacture of DNA in the US is dominated by a few companies that work closely with the government. By contrast, the technology used to build and train AI models is more widespread. “You can’t put that genie back in the bottle,” says Clore. “If you have the resources to try to trick us into making a DNA sequence, you can probably train a large language model.”
technologyreview.com/2025/10…

2
4
16
3,417

Using its EvoDiff generative-protein model, a Microsoft red-team redesigned known toxins so their DNA blueprints no longer tripped the alarms of commercial gene-synthesis screeners yet software still predicted the molecules would remain lethal.
DNA-order screening is governments’ main choke-point to stop biothreats.
If AI can routinely mutate toxins just enough to slip past filters, that defence grows porous.
Microsoft notified U.S. officials and the four big screening-software vendors.
A patch went out, but researchers say some AI-altered toxins still evade detection so the fix is only partial and the “arms race” has begun.
What experts propose next:
Move from simple sequence block-lists to AI-driven “functional similarity” scoring.
Add refusal/watermark layers inside protein-design models to block or track dangerous exports.
Create a mandatory, universal audit trail for all DNA orders including desktop synthesizers so malicious requests can’t easily shop around.
The takeaway: generative AI is now powerful enough to discover novel bio-“jailbreaks,” forcing regulators and industry to rethink biosecurity from model training all the way to DNA printers.

2
11
1,222

31 Jul 2025
They're actually both ProtT5 -- gotta correct the EvoDiff preprint. In practice, you'll get very similar results using any reasonable PLM.
1
3
115

26 Jul 2025
Another question, in EvoDiff paper you guys used ProtT5, and here you use ProtBert for the FPD calculations.
I'm interested in using FPD myself but was wondering how to choose the pLM. Is there any reason you guys preferred ProtBert over other pLMs, or in your exp. all are ok?
1
4
483

13 Jul 2025
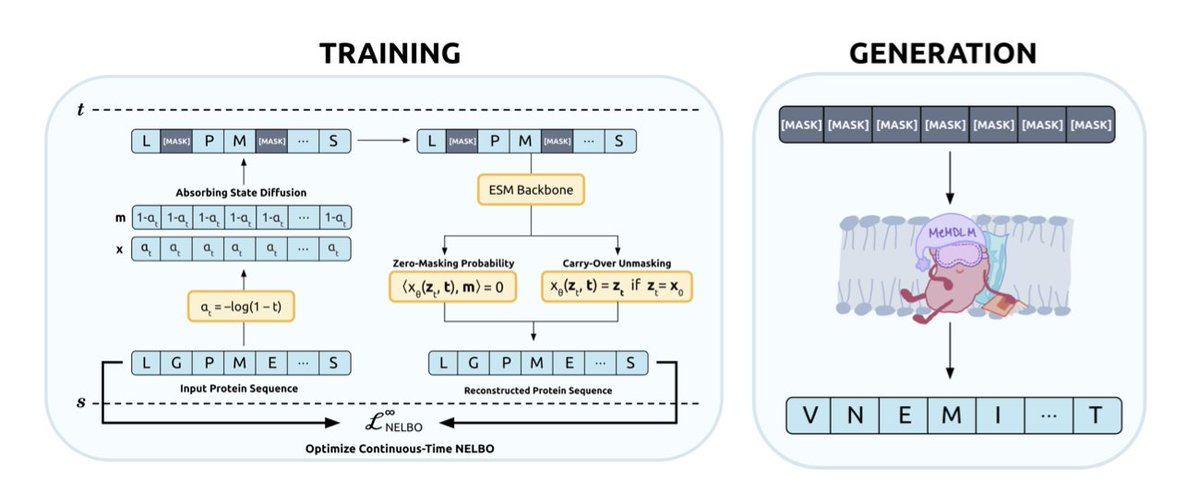
MEMDLM: De Novo MEMBRANE PROTEIN DESIGN WITH PROPERTY-GUIDED DISCRETE DIFFUSION
A new masked diffusion language model called MeMDLM has been developed for designing novel membrane proteins. This model utilizes the state-of-the-art protein language model ESM-2.
MeMDLM addresses challenges in membrane protein design by enabling parallel, non-sequential generation, which helps capture global sequence dependencies without requiring structural input. This is a significant advantage as high-resolution structural data for membrane proteins is limited.
The model outperforms autoregressive (AR) methods by generating sequences with enhanced transmembrane (TM) characteristics. It also effectively scaffolds soluble and TM motifs, showing greater biological similarity to original sequences compared to other inpainting methods.
MeMDLM incorporates a generalized Bayesian optimization procedure that uses saliency maps to guide the generation of soluble membrane proteins. This innovation facilitates potential experimental applications.
The training process involves pre-training ESM-2-150M on a broad range of protein sequences using the MDLM task, followed by fine-tuning with membrane proteins to specialize in de novo membrane protein sequence generation.
Evaluation results show that MeMDLM-generated proteins have a soluble residue density closely matching natural membrane proteins, indicating its ability to learn underlying distributions effectively.
Classifier-guided sampling is integrated with LaMBO-2, allowing for property-guided optimization of sequences for solubility and TM characteristics. This controllable discrete diffusion operates in a continuous latent space.
The method demonstrates that tokens with higher saliency scores are more likely to be optimized, particularly in insoluble regions, highlighting a direct link between continuous latent structure and discrete token optimization.
Visualizations of MeMDLM-generated sequences using AlphaFold3 confirm the presence of hallmark membrane protein structures, such as alpha-helical bundles and central TM regions.
MeMDLM-inpainted sequences achieve lower average pseudo-perplexities and maintain high cosine similarities compared to state-of-the-art inpainting methods like EvoDiff, preserving biological relevance while scaffolding functional motifs with greater confidence.
💻Code: github.com/kuleshov-group/md…
📜Paper: openreview.net/forum?id=ZnEx…
#MembraneProteinDesign #AIinBiology #ProteinEngineering #DeepLearning #ComputationalBiology
2
10
1,559
13 Jul 2025
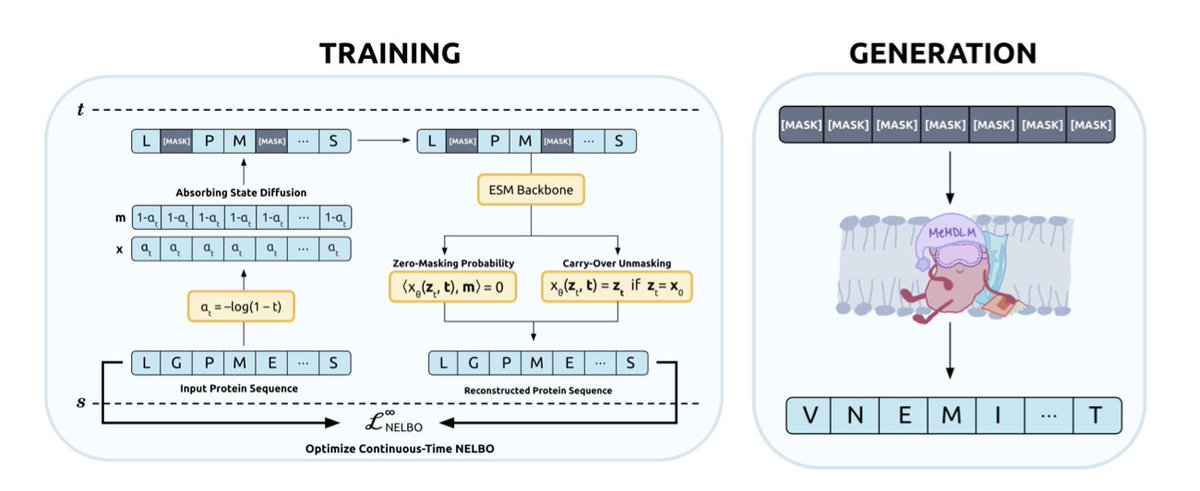
MEMDLM: De Novo MEMBRANE PROTEIN DESIGN WITH PROPERTY-GUIDED DISCRETE DIFFUSION
A new masked diffusion language model called MeMDLM has been developed for designing novel membrane proteins. This model utilizes the state-of-the-art protein language model ESM-2.
MeMDLM addresses challenges in membrane protein design by enabling parallel, non-sequential generation, which helps capture global sequence dependencies without requiring structural input. This is a significant advantage as high-resolution structural data for membrane proteins is limited.
The model outperforms autoregressive (AR) methods by generating sequences with enhanced transmembrane (TM) characteristics. It also effectively scaffolds soluble and TM motifs, showing greater biological similarity to original sequences compared to other inpainting methods.
MeMDLM incorporates a generalized Bayesian optimization procedure that uses saliency maps to guide the generation of soluble membrane proteins. This innovation facilitates potential experimental applications.
The training process involves pre-training ESM-2-150M on a broad range of protein sequences using the MDLM task, followed by fine-tuning with membrane proteins to specialize in de novo membrane protein sequence generation.
Evaluation results show that MeMDLM-generated proteins have a soluble residue density closely matching natural membrane proteins, indicating its ability to learn underlying distributions effectively.
Classifier-guided sampling is integrated with LaMBO-2, allowing for property-guided optimization of sequences for solubility and TM characteristics. This controllable discrete diffusion operates in a continuous latent space.
The method demonstrates that tokens with higher saliency scores are more likely to be optimized, particularly in insoluble regions, highlighting a direct link between continuous latent structure and discrete token optimization.
Visualizations of MeMDLM-generated sequences using AlphaFold3 confirm the presence of hallmark membrane protein structures, such as alpha-helical bundles and central TM regions.
MeMDLM-inpainted sequences achieve lower average pseudo-perplexities and maintain high cosine similarities compared to state-of-the-art inpainting methods like EvoDiff, preserving biological relevance while scaffolding functional motifs with greater confidence.
💻Code: github.com/kuleshov-group/md…
📜Paper: openreview.net/forum?id=ZnEx…
#MembraneProteinDesign #AIinBiology #ProteinEngineering #DeepLearning #ComputationalBiology
8
1,068
1 Jul 2025
PRO‐LDM: A Conditional Latent Diffusion Model for Protein Sequence Design and Functional Optimization
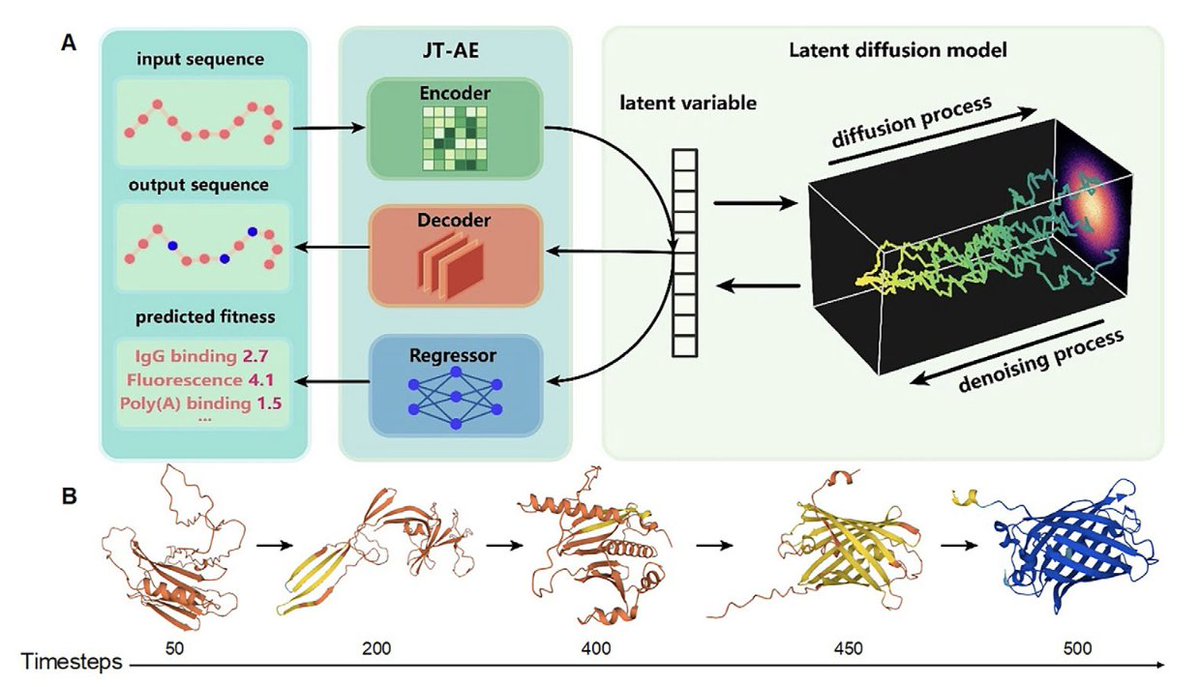
1.PRO-LDM introduces a modular latent diffusion model for full-length protein sequence design with both unconditional generation and functional optimization, combining accuracy with computational efficiency.
2.A major innovation lies in applying diffusion in the latent space, significantly reducing sampling cost while preserving the fidelity and diversity of generated sequences.
3.PRO-LDM achieves controllable design of protein sequences with target properties (e.g., fluorescence, solubility, thermal/chemical stability) by integrating conditional latent diffusion with supervised fitness prediction.
4.Outlier protein design is enabled by classifier-free guidance adjustment, allowing exploration of sequence space far beyond natural protein distributions, yet still producing foldable and functional variants.
5.The model successfully designed a new GFP variant, pro_2421, with 127× fluorescence intensity compared to wild-type GFP, plus enhanced solubility and thermal/chemical stability—verified both in silico and experimentally.
6.Compared to other state-of-the-art models like EvoDiff, ProteinMPNN, ESM3, and ProGen2, PRO-LDM produced more native-like sequences (lower KL divergence) and better foldability (higher pLDDT), with higher predicted fitness.
7.Joint training of encoder and diffusion modules outperforms frozen-language model embeddings (e.g., ESM2), yielding clearer latent-function mappings and better generalization for fitness prediction.
8.The model architecture is modular: swapping in pretrained encoders like ESM2 improves generalization, and training on diverse datasets (e.g., Swissprot, CATH) enables de novo generation of novel protein folds.
9.In unconditional mode, PRO-LDM matches or exceeds VAE and JT-AE in sequence identity and entropy profiles, capturing key evolutionary and biochemical properties without needing MSA alignment.
10.In conditional mode, the model accurately tunes fitness distributions across multiple datasets, generalizing across mutations, insertions/deletions, and sequence length variability.
11.Fitness prediction is performed jointly with sequence generation, allowing seamless integration of prediction and design. Regressor accuracy is on par with dedicated predictors.
12.Classifier-free guidance parameter ω controls diversity vs. fidelity tradeoff: ω ≈ 1 yields in-distribution high-fitness sequences; ω > 20 enables out-of-distribution exploration while maintaining foldability.
13.The model demonstrates broad generalization potential—when trained on CATH, it generated highly novel sequences with minimal similarity to training data yet retained foldability, outperforming EvoDiff in sequence divergence.
14.Experimental results confirm that PRO-LDM designed variants achieve functional improvements beyond training set benchmarks, validating its utility in real-world protein engineering.
📜Paper: advanced.onlinelibrary.wiley…
#ProteinDesign #DiffusionModel #SyntheticBiology #DeepLearning #ComputationalBiology
2
17
1,043
1 Jul 2025
PRO‐LDM: A Conditional Latent Diffusion Model for Protein Sequence Design and Functional Optimization
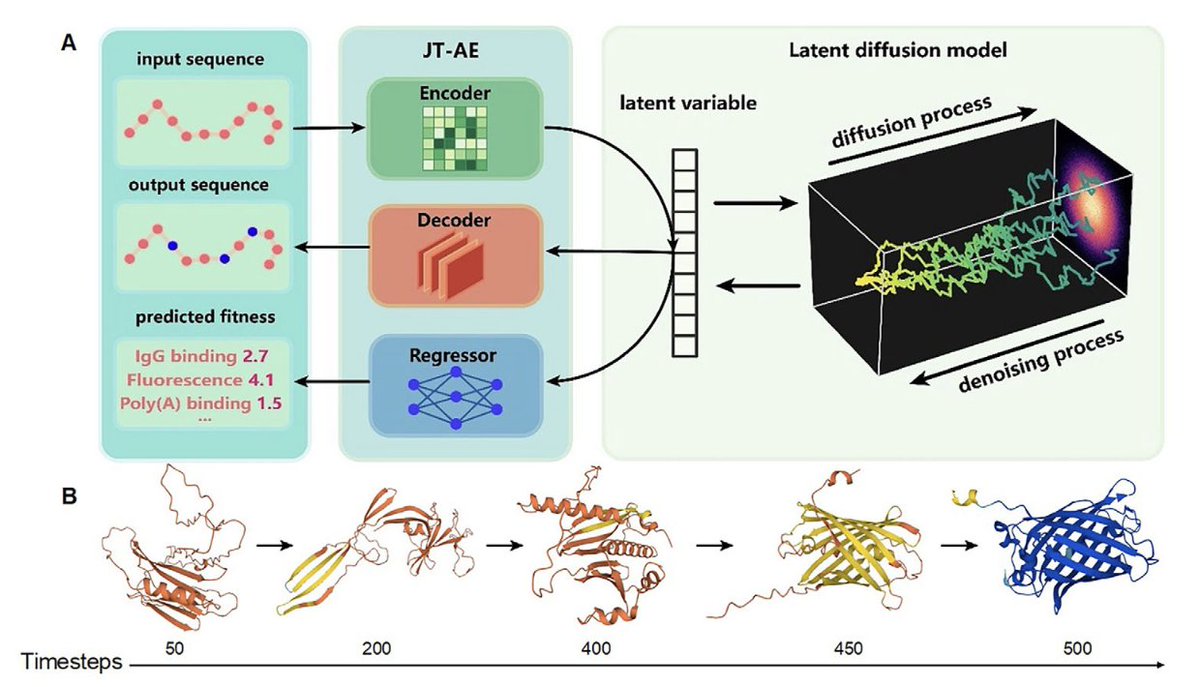
1.PRO-LDM introduces a modular latent diffusion model for full-length protein sequence design with both unconditional generation and functional optimization, combining accuracy with computational efficiency.
2.A major innovation lies in applying diffusion in the latent space, significantly reducing sampling cost while preserving the fidelity and diversity of generated sequences.
3.PRO-LDM achieves controllable design of protein sequences with target properties (e.g., fluorescence, solubility, thermal/chemical stability) by integrating conditional latent diffusion with supervised fitness prediction.
4.Outlier protein design is enabled by classifier-free guidance adjustment, allowing exploration of sequence space far beyond natural protein distributions, yet still producing foldable and functional variants.
5.The model successfully designed a new GFP variant, pro_2421, with 127× fluorescence intensity compared to wild-type GFP, plus enhanced solubility and thermal/chemical stability—verified both in silico and experimentally.
6.Compared to other state-of-the-art models like EvoDiff, ProteinMPNN, ESM3, and ProGen2, PRO-LDM produced more native-like sequences (lower KL divergence) and better foldability (higher pLDDT), with higher predicted fitness.
7.Joint training of encoder and diffusion modules outperforms frozen-language model embeddings (e.g., ESM2), yielding clearer latent-function mappings and better generalization for fitness prediction.
8.The model architecture is modular: swapping in pretrained encoders like ESM2 improves generalization, and training on diverse datasets (e.g., Swissprot, CATH) enables de novo generation of novel protein folds.
9.In unconditional mode, PRO-LDM matches or exceeds VAE and JT-AE in sequence identity and entropy profiles, capturing key evolutionary and biochemical properties without needing MSA alignment.
10.In conditional mode, the model accurately tunes fitness distributions across multiple datasets, generalizing across mutations, insertions/deletions, and sequence length variability.
11.Fitness prediction is performed jointly with sequence generation, allowing seamless integration of prediction and design. Regressor accuracy is on par with dedicated predictors.
12.Classifier-free guidance parameter ω controls diversity vs. fidelity tradeoff: ω ≈ 1 yields in-distribution high-fitness sequences; ω > 20 enables out-of-distribution exploration while maintaining foldability.
13.The model demonstrates broad generalization potential—when trained on CATH, it generated highly novel sequences with minimal similarity to training data yet retained foldability, outperforming EvoDiff in sequence divergence.
14.Experimental results confirm that PRO-LDM designed variants achieve functional improvements beyond training set benchmarks, validating its utility in real-world protein engineering.
📜Paper: advanced.onlinelibrary.wiley…
#ProteinDesign #DiffusionModel #SyntheticBiology #DeepLearning #ComputationalBiology
4
24
2,163
17 Jun 2025
Artificial intelligence methods for protein folding and design @COSB_CRSB
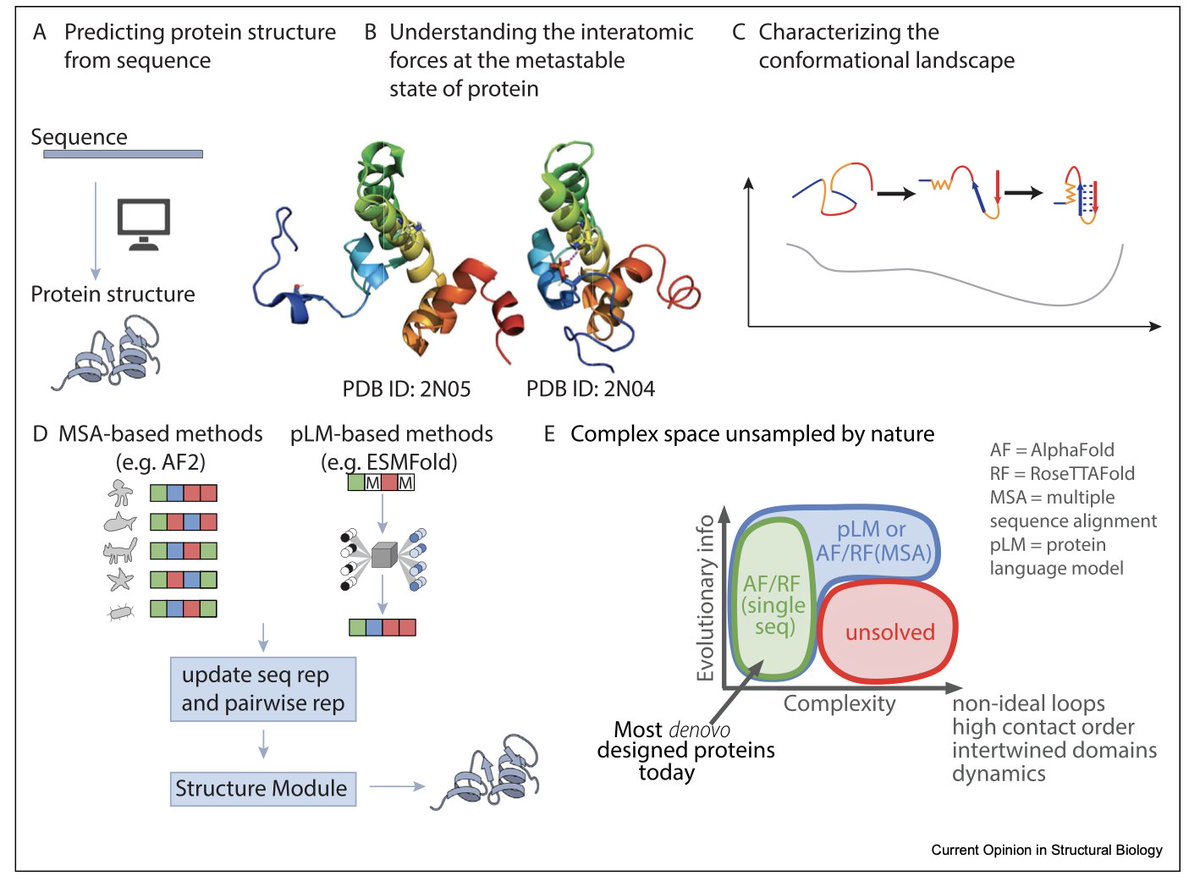
1.This review highlights a major paradigm shift: protein structure prediction models like AlphaFold2 (AF2) and RoseTTAFold have been successfully repurposed for de novo protein design. Innovations such as RFdiffusion and relaxed sequence optimization enable generative design of complex structures, pushing the boundary of what’s possible in protein engineering.
2.Despite their accuracy, current structure prediction models still don’t understand the underlying physics of folding. Their reliance on evolutionary information (via MSAs or learned pLM parameters) means they often fail with proteins lacking homologs or having complex folding dynamics.
3.A key limitation: most models can’t handle conformational heterogeneity. Proteins are dynamic, but prediction tools are biased toward stable, crystallizable conformations. Capturing conformational landscapes remains a fundamental challenge for structure-based design and function inference.
4.The review distinguishes two design goals: "structure-based sequence design" (maximize P(sequence|structure)) versus true "inverse folding" (find a sequence whose lowest-energy state is the target structure). Most current methods, like ProteinMPNN and ESM-IF, solve only the former.
5.Common designability metrics (e.g., AlphaFold’s single-sequence mode) often fail to validate natural proteins and struggle with long sequences. This undermines their reliability in high-complexity designs, highlighting a need for more physics-aware validation tools.
6.A major open problem: sequence design models (e.g. EvoDiff, Progen2, Raygun) and structure-based models both tend to recombine existing sequence or structural motifs rather than explore truly novel spaces. Current evaluations of novelty, diversity, and complexity need to reflect this.
7.Emerging methods like BioEmu and Distributional Graphormer aim to capture long-timescale protein dynamics by modeling energy landscapes, but are limited by simulation data scarcity and database biases. Mischaracterization of conformational equilibria still occurs.
8.CryoDRGN, NMR, and molecular simulations are valuable experimental methods that can complement machine learning. The review suggests integrating such data into learning frameworks is essential to improve conformational modeling and guide functional protein design.
9.Interestingly, the review questions whether AlphaFold2’s folding pathway predictions actually reflect understanding of dynamics or are simply artifacts of its fragment-pairing strategy. There's still little evidence that these models “understand” protein folding mechanisms.
10.The review ends with a vision for unified models that go beyond static structure prediction to capture the full energy landscape: predicting folding, alternate states, and designing sequences for desired conformational dynamics. Achieving this would transform both biology and biotechnology.
📜Paper: sciencedirect.com/science/ar…
#ProteinDesign #AlphaFold #AI #Bioinformatics #MachineLearning #StructuralBiology #GenerativeModels #ProteinFolding
3
19
94
10,308
17 Jun 2025
Artificial intelligence methods for protein folding and design @COSB_CRSB
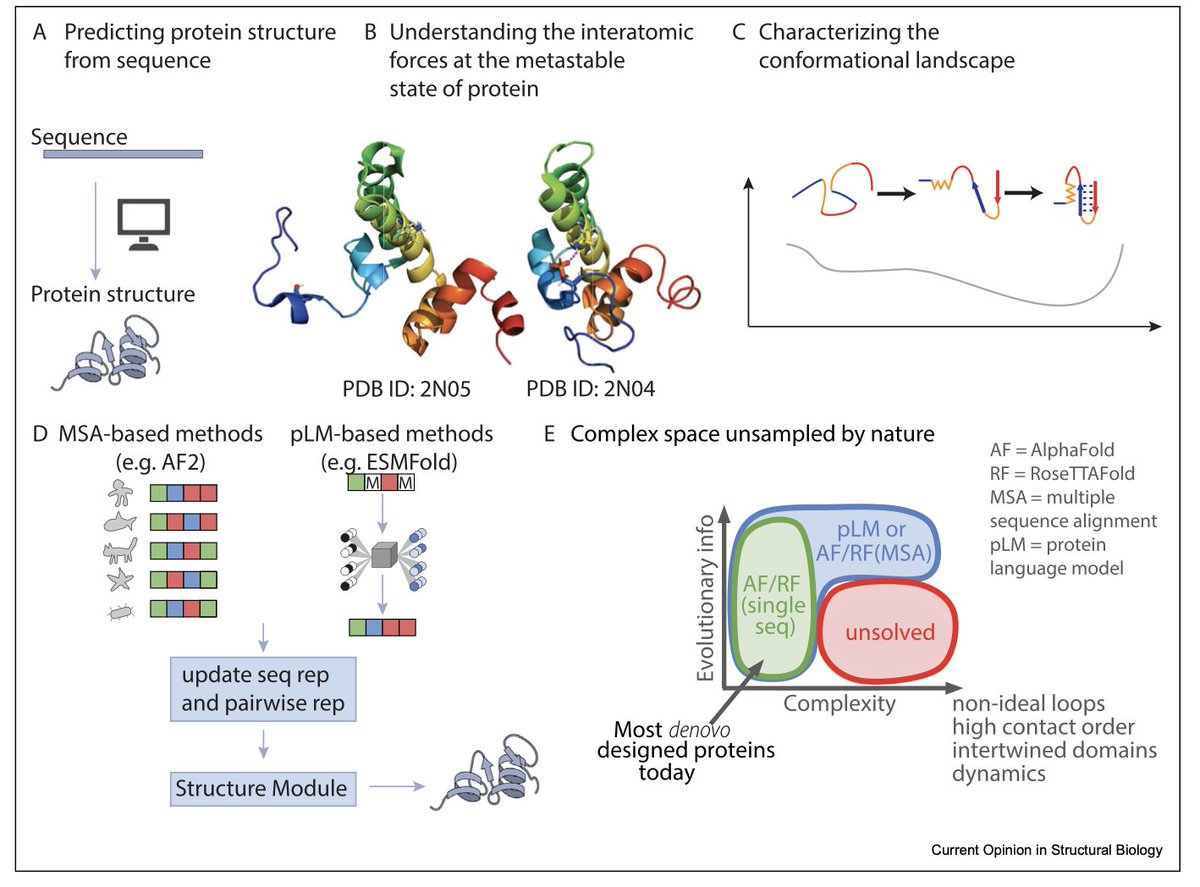
1.This review highlights a major paradigm shift: protein structure prediction models like AlphaFold2 (AF2) and RoseTTAFold have been successfully repurposed for de novo protein design. Innovations such as RFdiffusion and relaxed sequence optimization enable generative design of complex structures, pushing the boundary of what’s possible in protein engineering.
2.Despite their accuracy, current structure prediction models still don’t understand the underlying physics of folding. Their reliance on evolutionary information (via MSAs or learned pLM parameters) means they often fail with proteins lacking homologs or having complex folding dynamics.
3.A key limitation: most models can’t handle conformational heterogeneity. Proteins are dynamic, but prediction tools are biased toward stable, crystallizable conformations. Capturing conformational landscapes remains a fundamental challenge for structure-based design and function inference.
4.The review distinguishes two design goals: "structure-based sequence design" (maximize P(sequence|structure)) versus true "inverse folding" (find a sequence whose lowest-energy state is the target structure). Most current methods, like ProteinMPNN and ESM-IF, solve only the former.
5.Common designability metrics (e.g., AlphaFold’s single-sequence mode) often fail to validate natural proteins and struggle with long sequences. This undermines their reliability in high-complexity designs, highlighting a need for more physics-aware validation tools.
6.A major open problem: sequence design models (e.g. EvoDiff, Progen2, Raygun) and structure-based models both tend to recombine existing sequence or structural motifs rather than explore truly novel spaces. Current evaluations of novelty, diversity, and complexity need to reflect this.
7.Emerging methods like BioEmu and Distributional Graphormer aim to capture long-timescale protein dynamics by modeling energy landscapes, but are limited by simulation data scarcity and database biases. Mischaracterization of conformational equilibria still occurs.
8.CryoDRGN, NMR, and molecular simulations are valuable experimental methods that can complement machine learning. The review suggests integrating such data into learning frameworks is essential to improve conformational modeling and guide functional protein design.
9.Interestingly, the review questions whether AlphaFold2’s folding pathway predictions actually reflect understanding of dynamics or are simply artifacts of its fragment-pairing strategy. There's still little evidence that these models “understand” protein folding mechanisms.
10.The review ends with a vision for unified models that go beyond static structure prediction to capture the full energy landscape: predicting folding, alternate states, and designing sequences for desired conformational dynamics. Achieving this would transform both biology and biotechnology.
📜Paper: sciencedirect.com/science/ar…
#ProteinDesign #AlphaFold #AI #Bioinformatics #MachineLearning #StructuralBiology #GenerativeModels #ProteinFolding
1
9
972

17 Jun 2025
Ever wondered why masked diffusion outperforms other types of discrete diffusion? (e.g for EvoDiff)
Alan figured out and then fixed it!
16 Jun 2025

There are many domain-specific noise processes for discrete diffusion, but masking dominates! Why? We show masking exploits a key property of discrete diffusion, which we use to unlock the potential of those structured processes and beat masking! @gruver_nate @andrewgwils 1/7
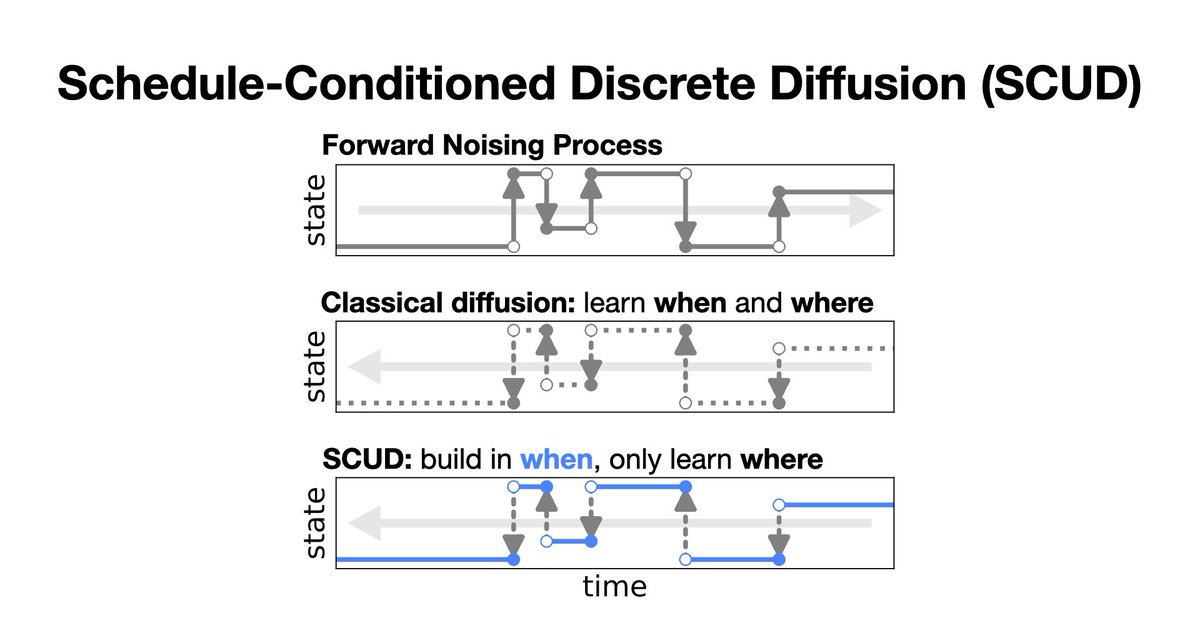
4
202
30 May 2025
ProSpero: Active Learning for Robust Protein Design Beyond Wild-Type Neighborhoods
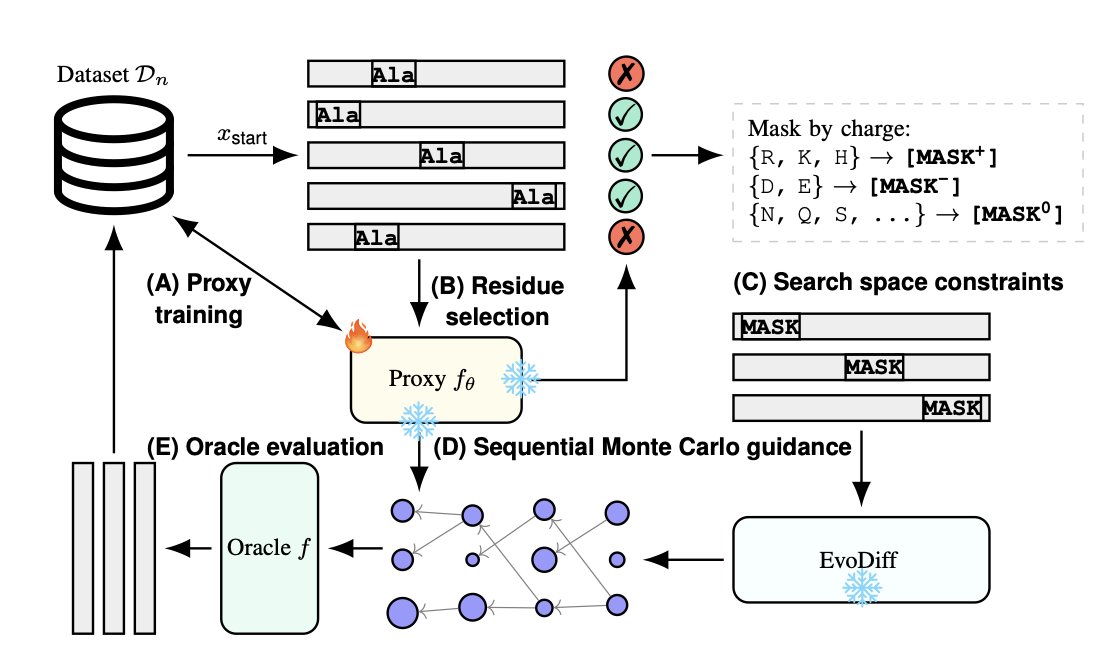
1.PROSPERO is a new framework for protein sequence design that actively guides a pre-trained generative model using a surrogate model updated via oracle feedback. Unlike many methods limited to local mutations near wild-type sequences, PROSPERO robustly explores novel regions while preserving biological plausibility.
2.The key innovation lies in inference-time guidance: instead of retraining generative models, PROSPERO leverages a frozen EvoDiff model and directs its outputs using an updated fitness proxy. This significantly reduces computational cost and overfitting risk.
3.PROSPERO introduces a targeted masking strategy that focuses mutations on fitness-relevant residues while protecting structurally critical ones. This strategy is inspired by alanine scanning and avoids disrupting conserved sites.
4.The second major innovation is biologically-constrained Sequential Monte Carlo (SMC) sampling. It restricts amino acid proposals to the same charge class as wild-type residues, ensuring that exploration remains within biologically plausible subspaces, even under proxy model misspecification.
5.Experiments on eight diverse protein engineering tasks show PROSPERO consistently achieves high-fitness sequences beyond the wild-type neighborhood. It ranks first on 5 tasks and second on 3 in terms of maximum fitness, outperforming methods like PEX, BO, and GFlowNets.
6.PROSPERO also breaks the typical trade-off between fitness and novelty. It retrieves sequences that are both highly fit and substantially different from the wild-type, often achieving 2–9× more novelty than methods like PEX with comparable fitness.
7.Diversity is maintained across top candidates, and the method produces sequences with strong folding confidence (pLDDT, pTM) and valid physicochemical properties, indicating biological plausibility.
8.Robustness tests under noisy surrogate models show PROSPERO outperforms baselines even when the proxy is unreliable. The advantage grows as the signal-to-noise ratio improves, highlighting its effective use of fitness signal.
9.Ablation studies confirm both targeted masking and biologically-constrained SMC are essential. Removing either leads to lower performance, especially in high-noise scenarios.
10.Overall, PROSPERO is a practical and general-purpose tool for protein design under limited data and expensive evaluations. It combines efficiency, robustness, and biological realism, making it a strong candidate for lab-in-the-loop applications.
📜Paper: arxiv.org/abs/2505.22494v1
#ProteinDesign #MachineLearning #Bioinformatics #ActiveLearning #ProteinEngineering #SyntheticBiology
5
19
1,483