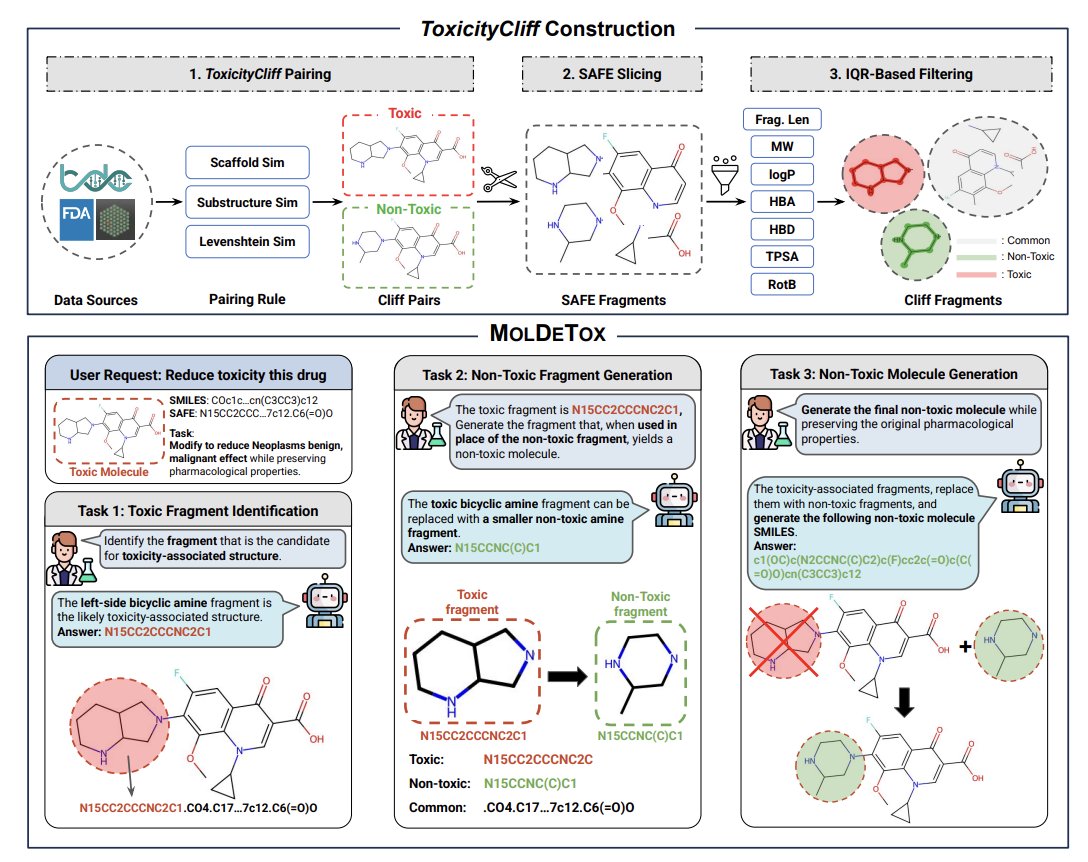
MolDeTox: Evaluating Language Model’s Stepwise Fragment Editing for Molecular Detoxification
1 MolDeTox introduces a stepwise benchmark that tests whether LLMs/VLMs can detoxify molecules via minimal, localized edits: identify toxicity-relevant fragments, propose non-toxic replacements, then generate a full non-toxic analog while preserving physicochemical properties.
2 The benchmark is built on a new dataset, ToxicityCliff: ~52,885 toxic/non-toxic molecule pairs across 49 toxicity endpoints, curated so each pair is globally similar but differs locally (toxicity label flips with small structural changes), reflecting realistic “design-around-toxicity” scenarios.
3 A key design choice is using SAFE, a fragment-level molecular representation (BRICS-based) that makes substructures explicit. MolDeTox uses SAFE both for interpreting molecules (fragment identification) and for generation (generate SAFE then decode to SMILES), aiming to reduce invalid structures and improve edit locality.
4 MolDeTox decomposes detoxification into three QA tasks with single-step vs multi-step variants: Task 1 toxic fragment identification (Ft-only), Task 2 non-toxic fragment generation (Fnt-only), Task 3 full non-toxic molecule generation (Mnt). This makes failures diagnosable rather than only scoring end-to-end success.
5 Data construction emphasizes “minimal-edit property preservation”: candidate pairs are selected with high similarity thresholds (scaffold/ECFP4/Levenshtein), then filtered with IQR-based rules to remove outliers in fragment counts/lengths and in key RDKit properties (MW, logP, TPSA, HBD, HBA, RotB).
6 Compared with prior detoxification benchmarks that often rely on proxy toxicity predictors, MolDeTox evaluates by matching to real non-toxic counterparts from curated pairs, avoiding single-model toxicity scoring but making the task harder and more reflective of exact-edit requirements.
7 Results show a consistent difficulty ladder: Task 1 > Task 2 > Task 3. Even strong models struggle on full-molecule exact match (Task 3), especially in multi-step edits, highlighting that fragment-level reasoning does not reliably translate into correct end-to-end reconstruction.
8 In-context learning is the most reliable boost. For example, GPT-5.2 with 4-shot improves Task 1 single-step accuracy (to ~54%) and substantially increases Task 3 single-step exact match under SAFE generation (to ~15.6%), indicating models benefit from retrieved, structurally similar examples.
9 SAFE-based generation improves chemical validity and property retention versus direct SMILES generation across models: reported gains include higher fingerprint similarity, higher validity, and higher PRS (Property Retention Score), supporting the idea that fragment-first generation better preserves the unchanged parts of a molecule.
10 The paper adds step-dependency analysis (8 outcome cases across T1/T2/T3 correctness), showing most Task 3 failures are “complete breakdown” (T1=0,T2=0,T3=0), while successes usually coincide with correct intermediate steps—evidence that the decomposition is informative for diagnosing where detoxification pipelines fail.
💻Code: github.com/datamol-io/safe
📜Paper: arxiv.org/abs/2605.12181
#ComputationalBiology #Cheminformatics #LLM #MolecularOptimization #DrugDiscovery #Toxicology #Benchmark #MultimodalAI #GenerativeAI #FragmentBasedDesign
8
22
1,669

5 Jul 2025
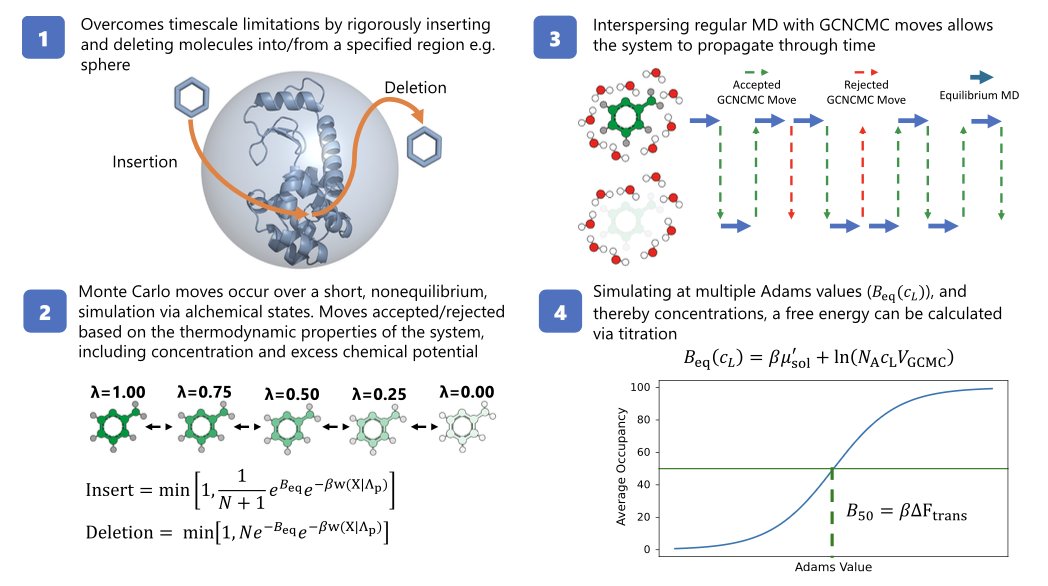
Accelerating fragment-based drug discovery using grand canonical nonequilibrium candidate Monte Carlo @NatureComms
1.This study introduces GCNCMC, a method that enables fragment insertion and deletion during MD simulations, accelerating fragment-based drug discovery by overcoming sampling challenges, especially for occluded binding sites and multiple binding modes.
2.Unlike traditional absolute binding free energy (ABFE) methods, GCNCMC does not require prior knowledge of binding poses, artificial restraints, or symmetry corrections. It samples multiple fragment binding modes naturally and ranks their binding affinities through titration-based simulations.
3.In head-to-head comparisons with classical mixed-solvent MD (MSMD), GCNCMC successfully identifies occluded binding sites in benchmark systems such as T4L99A and MUP1, which MSMD fails to detect even with extensive sampling.
4.The method applies nonequilibrium alchemical switches for ligand insertion/deletion, each governed by thermodynamic constraints, allowing for concentration-dependent acceptance of moves and accurate reconstruction of binding equilibrium.
5.GCNCMC reproduces known fragment concentrations in bulk water for systems like acetone and pyrimidine, validating its use in grand canonical ensemble simulations of small molecules beyond water.
6.In host–guest systems such as β-cyclodextrin, GCNCMC captures symmetric binding orientations without prior information, confirming the method’s ability to detect multiple binding modes and sample their correct populations.
7.For toluene binding to T4L99A, GCNCMC reproduces all four known binding modes with populations consistent with both ABFE and previously published data, using no a priori knowledge or transformation paths.
8.Binding affinities derived from GCNCMC titrations show high correlation with experimental data and conventional ABFE calculations. For 22 β-cyclodextrin fragments, the method achieved R² = 0.94 and τ = 0.84, indicating excellent predictive performance.
9.GCNCMC shows similar correlation with experiments for fragment binding to T4L99A and MUP1. While RMSE is slightly higher for protein systems due to added complexity, agreement with experimental and FEP-calculated affinities is strong.
10.Critically, GCNCMC avoids the need for multiple independent simulations per binding mode. A single titration simulation captures all populated modes, naturally weighting them by thermodynamics.
11.Limitations remain: GCNCMC may struggle with slow side-chain rearrangements or water displacement events during insertion/deletion steps. Future work will combine GCNCMC with other enhanced sampling tools like BLUES or FAST to address this.
12.Overall, this study establishes GCNCMC as a viable and efficient alternative to traditional free energy methods for fragment-based discovery, especially for targets with occluded pockets or complex binding geometries.
💻Code: github.com/essex-lab/grand-l…
📜Paper: nature.com/articles/s41467-0…
#ComputationalChemistry #DrugDiscovery #FragmentBasedDesign #MolecularDynamics #FreeEnergy #ProteinLigandBinding #GCNCMC #OpenMM

6
36
2,008
5 Jul 2025
Accelerating fragment-based drug discovery using grand canonical nonequilibrium candidate Monte Carlo @NatureComms
1.This study introduces GCNCMC, a method that enables fragment insertion and deletion during MD simulations, accelerating fragment-based drug discovery by overcoming sampling challenges, especially for occluded binding sites and multiple binding modes.
2.Unlike traditional absolute binding free energy (ABFE) methods, GCNCMC does not require prior knowledge of binding poses, artificial restraints, or symmetry corrections. It samples multiple fragment binding modes naturally and ranks their binding affinities through titration-based simulations.
3.In head-to-head comparisons with classical mixed-solvent MD (MSMD), GCNCMC successfully identifies occluded binding sites in benchmark systems such as T4L99A and MUP1, which MSMD fails to detect even with extensive sampling.
4.The method applies nonequilibrium alchemical switches for ligand insertion/deletion, each governed by thermodynamic constraints, allowing for concentration-dependent acceptance of moves and accurate reconstruction of binding equilibrium.
5.GCNCMC reproduces known fragment concentrations in bulk water for systems like acetone and pyrimidine, validating its use in grand canonical ensemble simulations of small molecules beyond water.
6.In host–guest systems such as β-cyclodextrin, GCNCMC captures symmetric binding orientations without prior information, confirming the method’s ability to detect multiple binding modes and sample their correct populations.
7.For toluene binding to T4L99A, GCNCMC reproduces all four known binding modes with populations consistent with both ABFE and previously published data, using no a priori knowledge or transformation paths.
8.Binding affinities derived from GCNCMC titrations show high correlation with experimental data and conventional ABFE calculations. For 22 β-cyclodextrin fragments, the method achieved R² = 0.94 and τ = 0.84, indicating excellent predictive performance.
9.GCNCMC shows similar correlation with experiments for fragment binding to T4L99A and MUP1. While RMSE is slightly higher for protein systems due to added complexity, agreement with experimental and FEP-calculated affinities is strong.
10.Critically, GCNCMC avoids the need for multiple independent simulations per binding mode. A single titration simulation captures all populated modes, naturally weighting them by thermodynamics.
11.Limitations remain: GCNCMC may struggle with slow side-chain rearrangements or water displacement events during insertion/deletion steps. Future work will combine GCNCMC with other enhanced sampling tools like BLUES or FAST to address this.
12.Overall, this study establishes GCNCMC as a viable and efficient alternative to traditional free energy methods for fragment-based discovery, especially for targets with occluded pockets or complex binding geometries.
💻Code: github.com/essex-lab/grand-l…
📜Paper: nature.com/articles/s41467-0…
#ComputationalChemistry #DrugDiscovery #FragmentBasedDesign #MolecularDynamics #FreeEnergy #ProteinLigandBinding #GCNCMC #OpenMM
7
857
18 May 2025
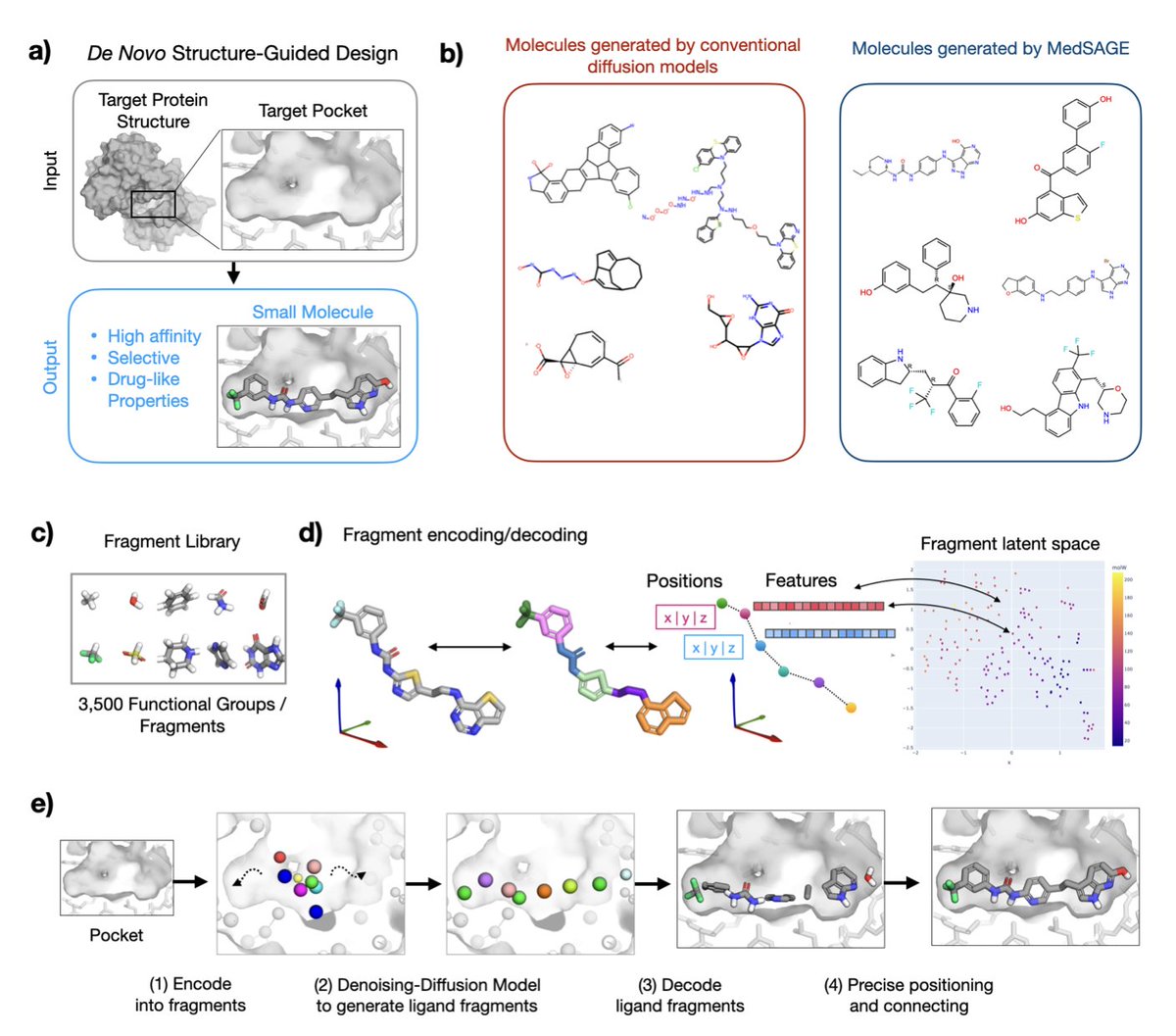
MedSAGE: Bridging Generative AI and Medicinal Chemistry for Structure-Based Design of Small Molecule Drugs
1.MedSAGE is a novel generative AI framework that addresses long-standing limitations in structure-based small-molecule drug design by operating directly on medicinal chemistry fragments instead of atoms or SMILES strings.
2.Unlike previous diffusion models that often generate chemically unstable or synthetically infeasible molecules, MedSAGE embeds functional groups and ring systems into a smooth latent space—improving interpretability, synthesizability, and design relevance.
3.The model employs a two-phase design: fragment generation via a 3D diffusion model guided by the protein pocket, followed by atomic-level assembly using a custom bond-connection algorithm that enforces chemical rules and optimizes Glide docking scores.
4.On a benchmark of 25 therapeutically relevant targets, MedSAGE-generated molecules had predicted affinities and selectivity statistically indistinguishable from those of approved drugs and clinical candidates—outperforming recent all-atom diffusion methods.
5.MedSAGE shows high selectivity: its ligands consistently reproduced more native binding interactions and avoided excessive hydrophobicity or bulkiness, unlike many molecules from DiffSBDD or IPDiff that lacked pocket specificity.
6.Despite no explicit optimization, MedSAGE molecules adhered to drug-likeness heuristics: Lipinski’s Rule of 5, appropriate logP, low synthetic complexity scores (~3.5), and scaffold diversity (~197 unique scaffolds out of 400 molecules per target).
7.Case studies showed MedSAGE could “rediscover” scaffolds structurally close to known binders like reboxetine, temsavir, and AK1, preserving key pharmacophores and even enhancing binding interactions via new hydrogen bonds.
8.Compared to traditional virtual screening of 30–400 million compound libraries, MedSAGE achieved comparable or better hit quality by generating only 2,000 molecules—offering a 100–1,000× improvement in hit enrichment efficiency.
9.Its fragment-based generation ensures aromatic ring planarity and reduces stereocenter complexity, avoiding common issues in atom-wise models. Fragment embeddings were learned via t-SNE and encode both 3D and chemical properties.
10.Although MedSAGE doesn’t yet handle protein flexibility or apo structures, it performs robustly on holo structure-based tasks—a realistic setting for many early-stage drug discovery projects.
11.The study introduces a scalable pipeline to pair AI-based molecule generation with similarity searches in large commercial libraries (e.g., Enamine REALSpace) to find purchasable analogs with similar binding profiles.
12.MedSAGE provides a compelling proof of concept that generative AI can capture medicinal chemistry principles and generate practical drug-like compounds directly from protein structures, with high interpretability and minimal data.
📜Paper: biorxiv.org/content/10.1101/…
#MedSAGE #DrugDesign #GenerativeAI #DiffusionModels #MedicinalChemistry #ProteinLigand #StructureBasedDesign #MolecularGeneration #FragmentBasedDesign #AI4Science #DeNovoDrugs #VirtualScreening #Bioinformatics #MachineLearning
1
18
76
5,721
18 May 2025
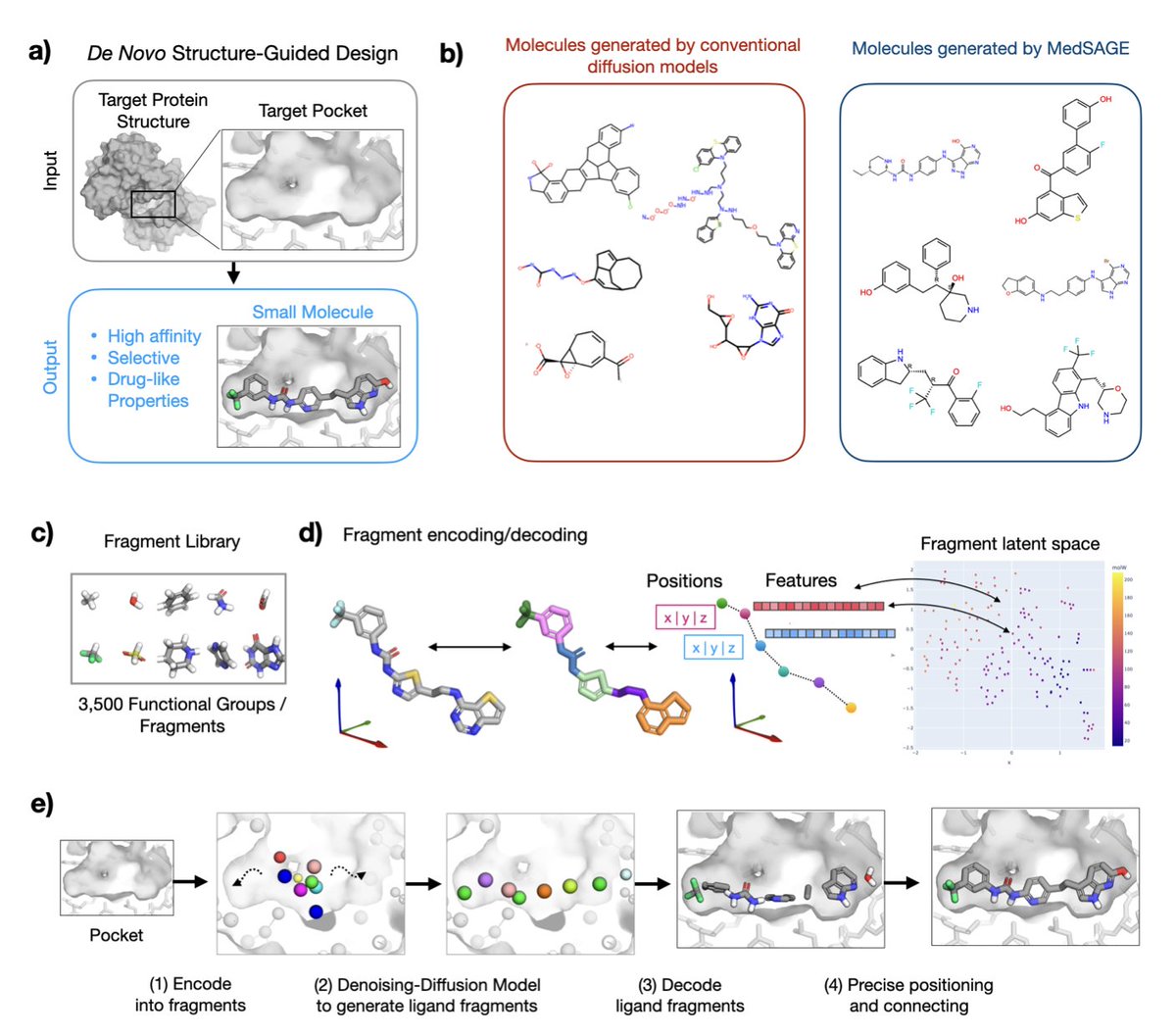
MedSAGE: Bridging Generative AI and Medicinal Chemistry for Structure-Based Design of Small Molecule Drugs
1.MedSAGE is a novel generative AI framework that addresses long-standing limitations in structure-based small-molecule drug design by operating directly on medicinal chemistry fragments instead of atoms or SMILES strings.
2.Unlike previous diffusion models that often generate chemically unstable or synthetically infeasible molecules, MedSAGE embeds functional groups and ring systems into a smooth latent space—improving interpretability, synthesizability, and design relevance.
3.The model employs a two-phase design: fragment generation via a 3D diffusion model guided by the protein pocket, followed by atomic-level assembly using a custom bond-connection algorithm that enforces chemical rules and optimizes Glide docking scores.
4.On a benchmark of 25 therapeutically relevant targets, MedSAGE-generated molecules had predicted affinities and selectivity statistically indistinguishable from those of approved drugs and clinical candidates—outperforming recent all-atom diffusion methods.
5.MedSAGE shows high selectivity: its ligands consistently reproduced more native binding interactions and avoided excessive hydrophobicity or bulkiness, unlike many molecules from DiffSBDD or IPDiff that lacked pocket specificity.
6.Despite no explicit optimization, MedSAGE molecules adhered to drug-likeness heuristics: Lipinski’s Rule of 5, appropriate logP, low synthetic complexity scores (~3.5), and scaffold diversity (~197 unique scaffolds out of 400 molecules per target).
7.Case studies showed MedSAGE could “rediscover” scaffolds structurally close to known binders like reboxetine, temsavir, and AK1, preserving key pharmacophores and even enhancing binding interactions via new hydrogen bonds.
8.Compared to traditional virtual screening of 30–400 million compound libraries, MedSAGE achieved comparable or better hit quality by generating only 2,000 molecules—offering a 100–1,000× improvement in hit enrichment efficiency.
9.Its fragment-based generation ensures aromatic ring planarity and reduces stereocenter complexity, avoiding common issues in atom-wise models. Fragment embeddings were learned via t-SNE and encode both 3D and chemical properties.
10.Although MedSAGE doesn’t yet handle protein flexibility or apo structures, it performs robustly on holo structure-based tasks—a realistic setting for many early-stage drug discovery projects.
11.The study introduces a scalable pipeline to pair AI-based molecule generation with similarity searches in large commercial libraries (e.g., Enamine REALSpace) to find purchasable analogs with similar binding profiles.
12.MedSAGE provides a compelling proof of concept that generative AI can capture medicinal chemistry principles and generate practical drug-like compounds directly from protein structures, with high interpretability and minimal data.
📜Paper: biorxiv.org/content/10.1101/…
#MedSAGE #DrugDesign #GenerativeAI #DiffusionModels #MedicinalChemistry #ProteinLigand #StructureBasedDesign #MolecularGeneration #FragmentBasedDesign #AI4Science #DeNovoDrugs #VirtualScreening #Bioinformatics #MachineLearning
1
4
868