
SpaceX Emerges as Major AI Infrastructure Player with Google Compute Deal
[BUSINESS]
SpaceX secures massive AI compute deal with Google.
Why it matters: SpaceX's substantial AI compute contract with Google fundamentally redefines its market position, shifting investor perception from solely an aerospace company to a critical infrastructure provider for artificial intelligence. This move highlights the increasing convergence of diverse industries in supporting the foundational needs of AI development, signaling a new competitive landscape for compute resources.
Follow DailyAIWire for the full brief.
🤔 How will the entry of non-traditional players like SpaceX impact the competitive dynamics of the AI infrastructure market?
#SpaceX #AIInfrastructure #GoogleCloud #GPUComputing #TechInvestment
15
Paul retweeted

Mar 20
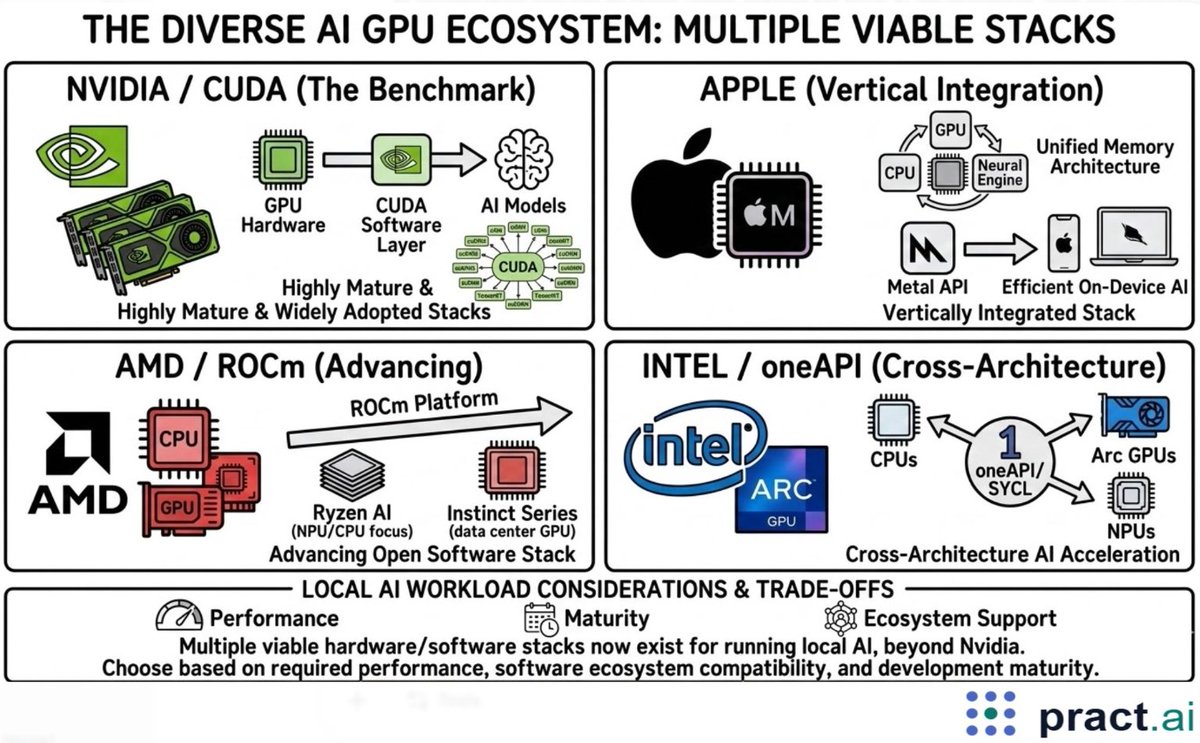
The current state of the GPU market for AI is more diverse than many assume
whyaiman.substack.com/p/the-…
#AI #GPUComputing #Llamacpp #localai #IntelArc
1
1
35

Jun 10
Snowball option pricing with GPU acceleration. DolphinDB Shark platform runs Monte Carlo simulations on NVIDIA GPUs—orders of magnitude faster than CPU-only approaches. #DolphinDB #GPUComputing #Derivatives
7

Megaport selects VAST Data to support AI data services across network and GPU platform... Read more: chiefit.me/megaport-selects-…
#ArtificialIntelligence #CloudInfrastructure #GPUComputing #HybridCloud #DataManagement #Multicloud @megaportnetwork @VAST_Data

15

Starting in 1 hour: The Future of Data-Driven Genomics
Join Complete Genomics, Google, and NVIDIA for a live webinar exploring how high-throughput sequencing, GPU-accelerated analysis, and AI-driven variant calling are enabling faster, more scalable genomics workflows.
The session will include a panel discussion with Biniam Feleke, Andrew Carroll, and Gary Burnett on how to connect sequencing, accelerated compute, and AI-enabled analysis.
⏰ 10:00 AM PT | 1:00 PM ET
Register here: na2.hubs.ly/H061YZX0
#Genomics #WholeGenomeSequencing #Bioinformatics #VariantCalling #AIinGenomics #GPUComputing #DeepVariant #NVIDIAParabricks #NGS

63

Jun 9
The ClearML team will be live at HPE Discover in Booth 2027!
Stop by to see how ClearML is helping enterprises scale AI infrastructure with GPU orchestration, multi-tenant AI environments, observability, workload automation, and AI infrastructure management built for real-world production teams.
Plus — swing by the booth for a chance to win the newest AI smart glasses. Would love to connect, talk AI infrastructure, and show you what’s next for enterprise AI operations.
More about HPE Discover:
hpe.com/us/en/discover/lasve…
#HPEDiscover #AIInfrastructure #MLOps #EnterpriseAI #GPUComputing #ClearML #AI @clearmlapp @HPE

53

Jun 8
What you’re looking at is not just another AI concept.
This is the vision behind the next generation of Synapse Power AI infrastructure — designed around the same principles used by leading technology companies building the future of artificial intelligence.
As AI adoption accelerates worldwide, one challenge continues to grow:
⚡ Massive power demand from GPUs.
Modern AI clusters can consume enormous amounts of electricity, while workloads often create sudden spikes in power usage that traditional infrastructure struggles to manage efficiently.
The future of AI is not only about more GPUs.
It’s about smarter power management, energy efficiency, grid stability, and infrastructure that can scale sustainably.
At Synapse Power, we continue to explore technologies and architectures that support the next era of AI computing — combining high-performance GPU infrastructure with intelligent energy optimization.
The future belongs to platforms that can deliver both compute power and operational efficiency.
🚀 Building the foundation for tomorrow’s AI economy.
#SynapsePower #AIInfrastructure #DataCenter #ArtificialIntelligence #GPUComputing #FutureOfAI #DigitalInfrastructure #AIEcosystem

2
2
4
76
Happening tomorrow: The Future of Data-Driven Genomics
Join Complete Genomics, Google, and NVIDIA for a webinar on how high-throughput sequencing, GPU-accelerated analysis, and AI-driven variant calling are helping researchers move from FASTQ files to accurate insights faster.
The session will feature a live panel discussion with Biniam Feleke, Andrew Carroll, and Gary Burnett on how to connect sequencing, accelerated compute, and AI-enabled analysis to enable more scalable genomics workflows.
📅 Tuesday, June 9
⏰ 10:00 AM PT | 1:00 PM ET
Register here: na2.hubs.ly/H061jl00
#Genomics #WholeGenomeSequencing #Bioinformatics #VariantCalling #AIinGenomics #GPUComputing #DeepVariant #NVIDIAParabricks #NGS

54

Jun 3
How do you build high-performance Julia code that runs across #CPUs and #GPUs without rewriting for every vendor? Join Oak Ridge National Laboratory scientists William F. Godoy and Philip Fackler for a live #webinar on JACC.jl, and see how vendor-neutral parallel computing in Julia can simplify HPC development while maintaining performance, portability, and developer productivity. juliahub.com/events/build-po…
#JuliaLang #HPC #GPUComputing #ParallelComputing #ScientificComputing
2
10
854
Jun 3
Exciting to see the collaboration between @clearmlapp and @armada_ai continue to grow.
Together, we’re helping power next-gen AI infrastructure with scalable multi-tenant environments, GPU optimization, orchestration, and AI factory services built for enterprise and sovereign AI deployments.
Love seeing innovative teams come together to help operators move beyond bare metal and accelerate real-world AI adoption.
Learn more at : armada.ai/blog/bridge-market…
#AIInfrastructure #MLOps #GPUComputing #EnterpriseAI #ClearML #ArmadaAI

2
53

May 17
CME Group GPU Compute Futures: AI Infrastructure Hedging
weeklyreviewer.com/dive-deep…
#AIInfrastructure #GPUFutures #CMEFutures #AIHedging #ComputeTrading #FuturesMarket #FinTech #Derivatives #GPUComputing #AIFinance #ArtificialIntelligence #TechInvesting
14
692

In a world hungry for computing power, @DeepBrainChain is the bridge.
By connecting idle GPUs into a seamless, decentralized network, we’re ensuring that the next big AI breakthrough isn't slowed down by hardware shortages.
#DBC #GPUComputing #DePIN #Blockchain

2
4
14
935

May 9
📢 RA 𝐎𝐩𝐩𝐨𝐫𝐭𝐮𝐧𝐢𝐭𝐲 𝐚𝐭 𝐈𝐈𝐓 (𝐈𝐒𝐌) 𝐃𝐡𝐚𝐧𝐛𝐚𝐝
Applications are invited for a Research Associate-I (Postdoctoral) position under an Anusandhan National Research Foundation (ANRF)-ARG sponsored project at the the Department of Physics, IIT (ISM) Dhanbad)
🔬 Project Title:
Strong Correlation, Entanglement and Complexity in Topological and Flat Band Systems: A GPU Accelerated Approach
The project focuses on cutting-edge problems in:
✔️ Strongly correlated quantum systems
✔️ Topological and flat-band physics
✔️ Quantum information inspired approaches
✔️ GPU-accelerated computational many-body methods
✔️ Entanglement and dynamical mean-field techniques
We are looking for motivated researchers with strong background in:
• Theoretical/Computational Condensed Matter Physics
• Quantum many-body physics
• Programming and scientific computing
• Strongly correlated or topological systems
💰 Fellowship: ₹58,000/month HRA
📍 Location: Dhanbad, India
📅 Last date to apply: 18 May 2026
Interested candidates should send their CV, research proposal, past research experience, and publication list (single PDF) to:
📧 ra1topology@gmail.com
To know more about Prof. Sen's research group visit: sites.google.com/view/sudesh…
#Postdoc #ResearchAssociate #CondensedMatterPhysics #QuantumPhysics #TopologicalPhysics #StronglyCorrelatedSystems #QuantumInformation #GPUComputing #ComputationalPhysics #ANRF #IITISM #PhysicsResearch
1
9
521

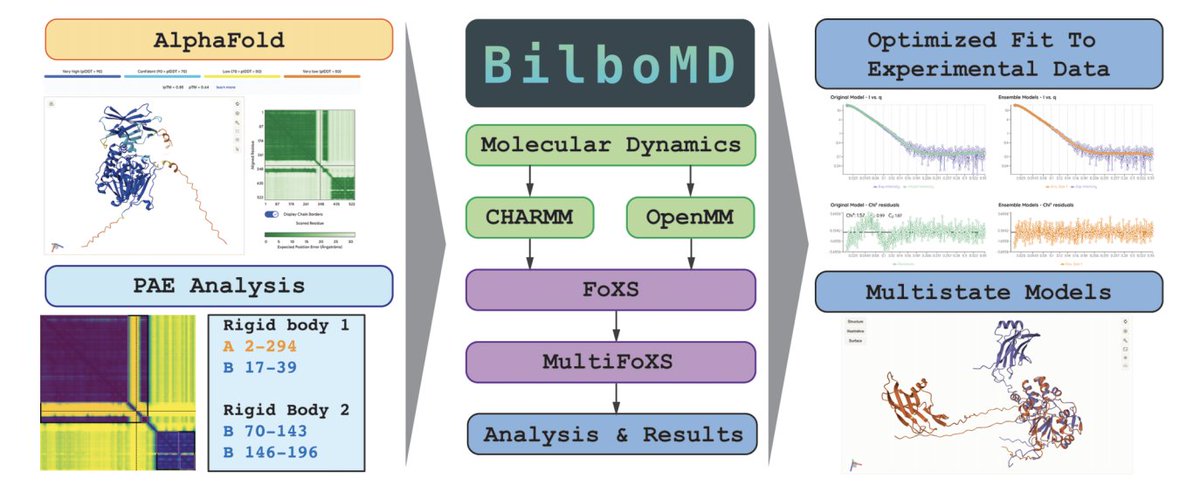
BilboMD: A web-accessible SAXS and AlphaFold-guided modeling pipeline
1. BilboMD is a free, no-login web platform for integrative modeling of flexible macromolecules using experimental SAXS/SANS plus atomistic starting models (experimental structures or AI predictions), aiming to make ensemble-based solution modeling accessible and reproducible.
2. A key innovation is automated definition of rigid vs flexible regions from AlphaFold confidence outputs: the PAE Jiffy converts AlphaFold PAE matrices (plus pLDDT) into CHARMM-compatible constraints, using Leiden clustering on a residue-residue similarity graph, then outputs a deterministic const.inp users can review and tweak.
3. BilboMD couples constrained conformational sampling (MD) with scattering-profile fitting: it generates many conformers, computes theoretical SAXS curves with FoXS, and selects best single- and multi-state ensembles with MultiFoXS—explicitly targeting systems where multiple conformations coexist in solution.
4. The pipeline uses a reduced MD representation: rigid domains are treated as frozen bodies while flexible linkers/regions are simulated, dramatically shrinking degrees of freedom so more conformational space can be explored for SAXS-consistent ensembles.
5. BilboMD automatically derives SAXS-driven radius-of-gyration restraints via a comprehensive Guinier scan across q-windows (with validation criteria like R² thresholds and qRg bounds), then biases MD sampling with harmonic restraints centered on the experimental Rg.
6. GPU acceleration is a major practical advance: an OpenMM-based variant (deployed on NERSC) delivers large speedups over CPU-based CHARMM for the MD step (reported 18×, 31×, and 78× on test systems), enabling larger conformer pools and more extensive ensemble fitting in feasible wall time.
7. Usability features target non-specialists while supporting power users: browser-based job submission with validation and sensible defaults, interactive Inp Jiffy for manual constraint authoring, results pages with before/after fits, residuals, downloadable bundles, and embedded Mol* visualization; API access supports automation and workflow integration.
8. BilboMD includes a diagnostic “feedback report” that goes beyond a final χ²: it estimates an appropriate ensemble state count (stopping when χ² improvement is no longer meaningful), checks molecular weight consistency (Vc-based MW) to flag aggregation/oligomer mismatch, and interprets fit failures by q-range to suggest concrete constraint edits.
9. Case studies illustrate typical outcomes: (i) Staphylococcal Eap required a 2-state ensemble to fit SAXS well (χ² improved from ~1.94 to ~1.17), reflecting domain flexibility; (ii) a PP1/PTG complex achieved a strong 2-state fit (χ² ~0.98); (iii) an aggregated sample was correctly diagnosed as not fixable by modeling because SAXS MW indicated major aggregation.
💻Code: github.com/bl1231/bilbomd
📜Paper: doi.org/10.1093/nar/gkag377
#SAXS #SANS #IntegrativeStructuralBiology #AlphaFold #MolecularDynamics #OpenMM #FoXS #EnsembleModeling #WebServer #ReproducibleResearch #HPC #GPUComputing
4
24
1,721

Apr 22
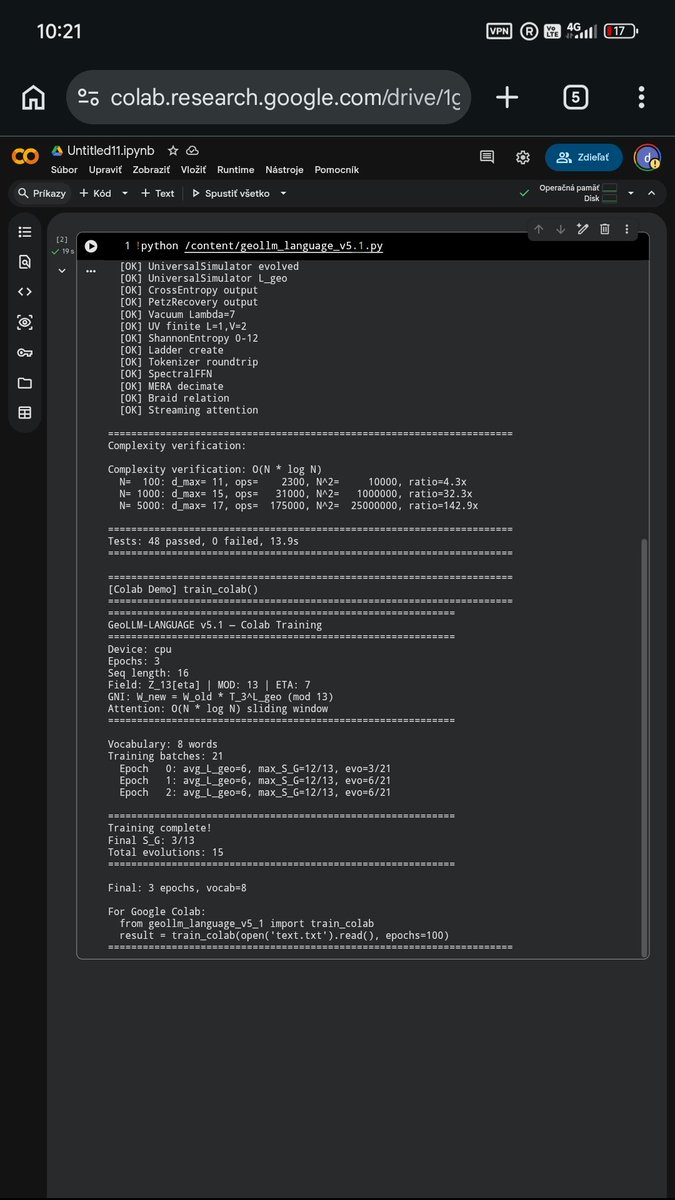
I finished working on GeoLLM v5.1. The architecture is now fully vectorized in native PyTorch, but runs without using floats (decimal numbers). The entire model operates exactly over the Z_13 integer array.
From today's hardware benchmarks:
▪️ Attention complexity confirmed at O(N log N). At 5,000 tokens, the network performs 175,000 operations instead of 25 million (standard Transformer).
▪️ Cognitive stability: The system measures its own thermodynamic entropy. In a run-time test, it autonomously expanded its phase space (Hensel's lift) 15 times to prevent hallucinations.
The transition from theoretical mathematics to real hardware is complete.
@OpenAI @xai
#GeoLLM #pAdelicAI
#AIresearch #LLM #PyTorch #MachineLearning #Mathematics #GPUcomputing #DeepTech


2
4
392

Apr 19
Performance-driven AI requires more than just API calls. It requires infrastructure. ⚙️
We’ve open-sourced a collection of implementations for:
↳ NVIDIA Warp: GPU-accelerated differentiable physics workflows.
↳ Elastic Vector DBs: Consistent hashing and live ring visualization.
↳ Semantic Caching: Reducing latency and overhead in RAG pipelines.
Deep dive into the source: github.com/Marktechpost/Othe…. .
#AIInfrastructure #Python #VectorDatabase #GPUComputing

1
7
15
571

Apr 15
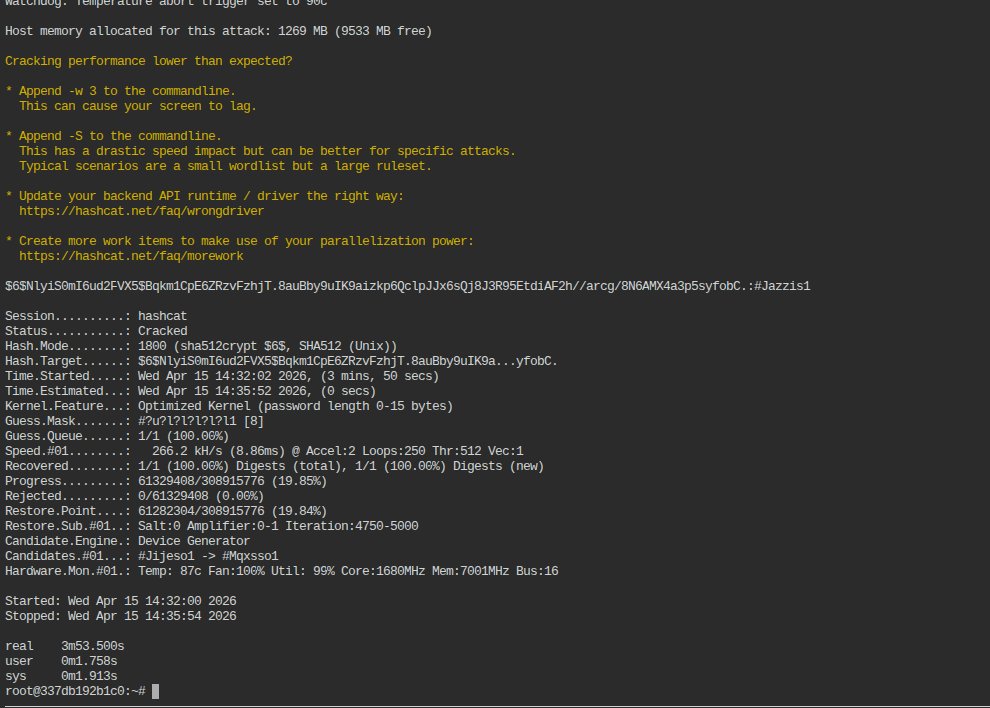
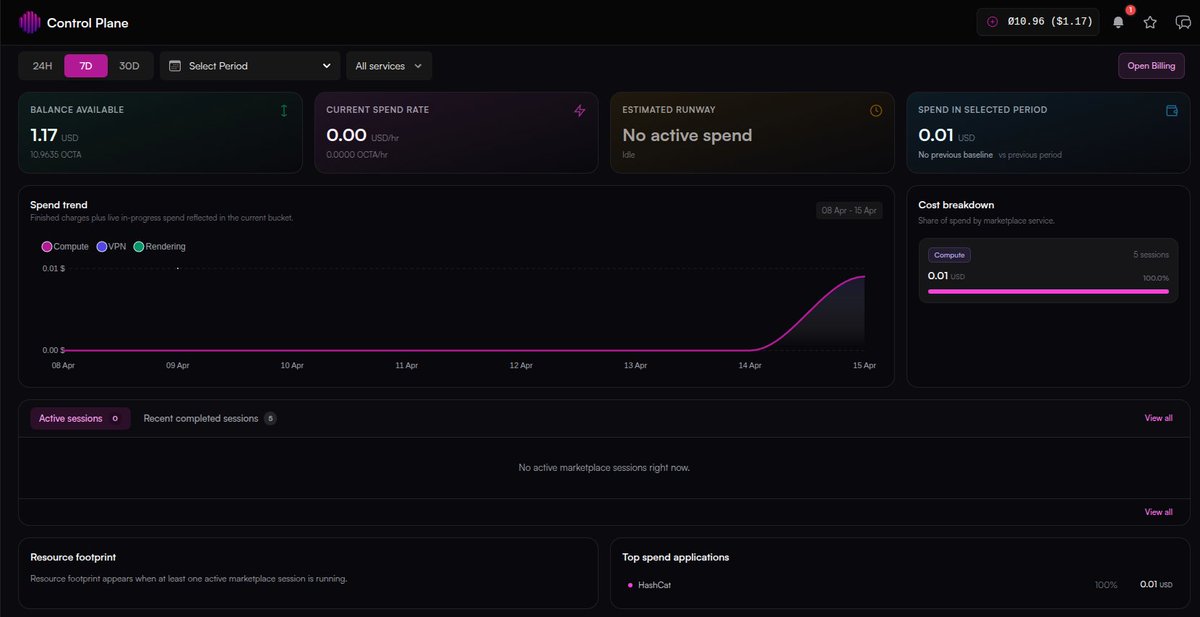
Game Over for the hashes! 💀
One of our team members had a hash that would've taken days to crack on his PC. We threw it onto #Octa and it was done in 3.53 minutes.
All that power for as low as $0.01. That's how we roll!
#HackTheBox #HTB #CyberSecurity #EthicalHacking #InfoSec #PenTesting #Crypto #OctaSpace #GPUComputing
6
19
63
4,839
Mar 23
This month in Julia world covers a lot of ground—from compiler and tooling progress to RayMakie, JETLS, GPU acceleration, new ecosystem releases, and developer experience improvements. A great snapshot of the ideas, tools, and momentum shaping the Julia community right now. discourse.julialang.org/t/th…
#JuliaLang #OpenSource #ScientificComputing #DeveloperTools #GPUComputing
1
7
661
Mar 20
Makie is taking a major step toward photorealistic scientific #visualization. With RayMakie and Hikari, researchers can turn existing Makie scenes into #GPU-accelerated ray-traced renders—bringing global illumination, volumetrics, and physically based materials directly into Julia workflows. makie.org/website/blogposts/…
#JuliaLang #Makie #ScientificVisualization #RayTracing #GPUComputing #ComputerGraphics #PathTracing #DataVisualization #ResearchComputing #HighPerformanceComputing
1
9
375
Mar 19
The intelligence revolution isn’t just about smarter models.
It’s about the power that allows them to train, scale, and operate in the real world.
Synapse Power stands behind this shift providing the compute that fuels the next generation of intelligent systems.
#FutureOfAI
#GPUComputing
#SynapsePowerAI

7
83