
Jun 9
🔬 What it does:
Pan-cancer ctDNA profiling from plasma
SNVs, indels, gene fusions & CNVs in one workflow
CHIP-aware: discriminates tumor variants from clonal hematopoiesis
UMI-based error correction panel of normals filtering
Dual variant calling with VarDict & Mutect2
1
46



Mar 15
From Grok SuperHeavy about Rosie:
The reported case is credible but involves significant collaboration with experts and established scientific workflows, rather than a fully autonomous AI-driven creation.
Paul Conyngham, a data and machine-learning expert with no formal biology training, sought treatment for his dog Rosie’s aggressive mast cell tumor after conventional therapies (surgery, chemotherapy, and immunotherapy) failed. He arranged whole-genome sequencing of healthy blood and tumor DNA through UNSW’s Ramaciotti Centre for Genomics (approximately AUD 3,000), generating raw data for analysis. Collaborating with UNSW researchers—including those at the RNA Institute—he designed and had manufactured a bespoke nanoparticle-formulated mRNA vaccine. Injections began around December 2025, and within weeks one tennis-ball-sized tumor reportedly shrank by roughly 50%, with improvements in energy and overall tumor burden noted in media accounts. Researchers involved have described the outcome as surprising and a potential “world-first” personalized mRNA cancer vaccine for a dog.
ChatGPT served primarily as an accessible research and coding assistant.** It helped Conyngham (who leveraged his existing computational skills) navigate literature, plan the sequencing request, interpret basic genomic workflows, and draft analysis scripts. This lowered the knowledge barrier for a non-specialist but did not replace professional bioinformatics or laboratory expertise.
AlphaFold played a supportive, downstream role in structural biology, not in identifying mutations themselves.** The mutations were detected through standard genomic comparison of tumor versus normal sequences using established variant-calling tools (e.g., those analogous to Mutect2 or similar pipelines). Once the mutated amino-acid sequences—particularly in the well-known canine mast cell tumor driver gene *c-KIT*—were identified, AlphaFold was used to predict the three-dimensional structure of the altered protein. This modeling visualized how specific mutations might disrupt folding, expose new surfaces, or create structural vulnerabilities. Such insights could inform epitope selection for the mRNA construct (i.e., which segments of the mutated protein to encode so that the immune system recognizes them) or help prioritize neoantigens. Reports note that one AlphaFold model had a relatively low confidence score (~54%), underscoring the need for experimental validation, as experts have cautioned. AlphaFold does not analyze raw genomic reads or “find” mutations; it operates on already-determined protein sequences. Its value here aligns with broader trends in immuno-oncology, where structural predictions increasingly refine neoantigen prioritization or peptide-MHC interaction modeling (capabilities expanded in AlphaFold3 and related tools).
Is this hype?** The core achievement is real and noteworthy: AI tools demonstrably accelerated a motivated individual’s ability to drive a personalized neoantigen-style mRNA vaccine in a veterinary setting, where regulatory hurdles are lower than in human medicine. Personalized mRNA vaccines targeting tumor-specific mutations are an established and actively researched modality (pursued by companies such as Moderna and BioNTech in human trials, with analogous work in canine oncology). A 50% tumor reduction is biologically plausible for a well-targeted neoantigen vaccine, especially if it elicited a strong T-cell response against driver mutations like those in *c-KIT*. However, the media framing—that ChatGPT and AlphaFold alone “created” a cure—overstates autonomy and underplays the critical contributions of professional sequencing, variant analysis, vaccine synthesis at a specialized institute, ethical approvals, and veterinary administration. The result remains a single anecdotal case without controls; mast cell tumors in dogs can show variable spontaneous or treatment-related responses, and longer-term data are unavailable.
1
3
73

Mar 14
That techbro knew that highlighting ChatGPT/ Alphafold will make it go viral - Catnip for X bros trying to monetize..
Bet interviewing the researcher involved will reveal how it was done
Per Grok -
### How mutations are usually identified from sequencing data (standard pipeline for neoantigens in personalized cancer vaccines)
The typical workflow for turning tumor sequencing into potential neoantigens (mutated peptides that could be vaccine targets) is a multi-step bioinformatics pipeline. This is done long before any structure prediction tool like AlphaFold comes into play. Here's the usual process in 2025–2026 clinical/research settings:
1. Sequencing:
- Whole-exome sequencing (WES), whole-genome sequencing (WGS), or targeted panels of tumor DNA.
- Matched normal (germline) DNA from blood/saliva for comparison.
- Often paired with tumor RNA-seq (to check expression of mutated genes).
2. Read alignment & pre-processing:
- Align raw reads to the human reference genome (e.g., using BWA, Bowtie2).
- Quality filtering, duplicate removal, etc.
3. Somatic mutation calling / variant detection (this is where mutations are actually identified):
- Compare tumor vs. normal to find somatic (tumor-specific) variants.
- Tools: Mutect2 (GATK), VarScan, Strelka2, MuTect, or ensemble callers for higher confidence.
- Focus on nonsynonymous mutations (missense, indels, frameshifts, fusions) that change the protein sequence.
- Filter for high variant allele frequency (VAF), expression in RNA-seq, clonal status, etc.
4. Translation to mutated peptides:
- Annotate variants with tools like VEP (Variant Effect Predictor) or ANNOVAR.
- Generate the altered protein sequence around the mutation (e.g., 21-mer or sliding windows).
- Split into possible epitope lengths (8–11 mers for MHC class I, longer for class II).
5. Neoantigen prediction & prioritization:
- Peptide-MHC binding affinity prediction (core step): NetMHCpan, MHCflurry, MixMHCpred → rank how well mutated peptides bind patient's HLA alleles (typed via sequencing or PCR).
- Additional filters: proteasomal cleavage (NetChop), TAP transport, expression level, clonality, dissimilarity to self (to avoid tolerance), immunogenicity scores.
- Pipelines: pVACtools, MuPeXI, nextNEOπ, NeoPredPipe, TruNeo, or commercial ones like those from BioNTech/Moderna/Genentech.
Only after this do you have a shortlist of candidate neoantigens (mutated peptides).
### Where AlphaFold actually fits in (limited role)
AlphaFold enters much later — and rarely as the main tool for pinpointing neoantigens:
- To model the 3D structure of a mutated protein or peptide-MHC complex → assess if the mutation dramatically alters folding, stability, or epitope presentation/conformation.
- To help understand why a neoantigen might be immunogenic (e.g., structural changes exposing new surfaces).
- In advanced vaccine design: refine peptide structures for better MHC binding or TCR interaction modeling (e.g., with tools like AlphaFold-Multimer or RoseTTAFold).
- In some emerging pipelines (post-2024): use AlphaFold structures as input features for improved immunogenicity predictors or to simulate neoantigen presentation.
But AlphaFold cannot replace variant calling or read sequencing data — it starts from an already-known mutated amino acid sequence. If someone claims "AlphaFold pinpointed the mutated proteins from genomic sequences," it's either loose wording (they mean used AlphaFold downstream after variant calling) or a misunderstanding of the tools.
In short: sequencing variant callers find the mutations → neoantigen pipelines predict peptides → AlphaFold (optionally) models structures for refinement. The heavy lifting for "identifying mutations" is done by dedicated genomic tools, not AlphaFold.
2
1
7
4,736

29 Oct 2025
This is different from local assembly like Mutect2 -- more like personalized whole genome assembly to create a better reference for each cancer (which is known to improve variant calls but is also a huge hassle)
2
200

17 Oct 2025
🧬 ~16% of humans die by Cancer globally and early detection can reduce this number hugely.
And Google's just released DeepSomatic - an open-source AI for cancer genetic analysis - speeds up the analysis by 10x.
detects cancer-causing mutations with far higher accuracy than existing tools.
DeepSomatic converts DNA read alignments into images and uses a convolutional neural network to classify whether each site is normal or tumor-specific, and it can still work without matched normal tissue.
It was trained and tested on the CASTLE dataset, which combines whole-genome data from 6 cancer cell lines across those 3 sequencing platforms.
In tests, DeepSomatic raised insertion and deletion detection F1 scores from 80% to 90% on Illumina and from below 50% to above 80% on PacBio, a major gain for hard-to-spot variants.
Across experiments it found 329,011 somatic variants covering all tested reference lines and preserved samples.
It consistently beat older tools like MuTect2, SomaticSniper, Strelka2, and ClairS.
It even identified mutations in cancers it had never seen before, including glioblastoma and pediatric leukemia, and found 10 new mutations in the leukemia sample.

2
5
26
4,373

12 Oct 2025
🚀 Learn somatic variant analysis in cancer genomics! Hands-on online workshop: QC, GATK4 Mutect2, R & VEP annotation. Go from raw data to actionable insights🧬📊27-29 April
physalia-courses.org/courses…
#CancerGenomics #SomaticVariants #Bioinformatics #PrecisionOncology #RStats
1
7
555

4 Aug 2025
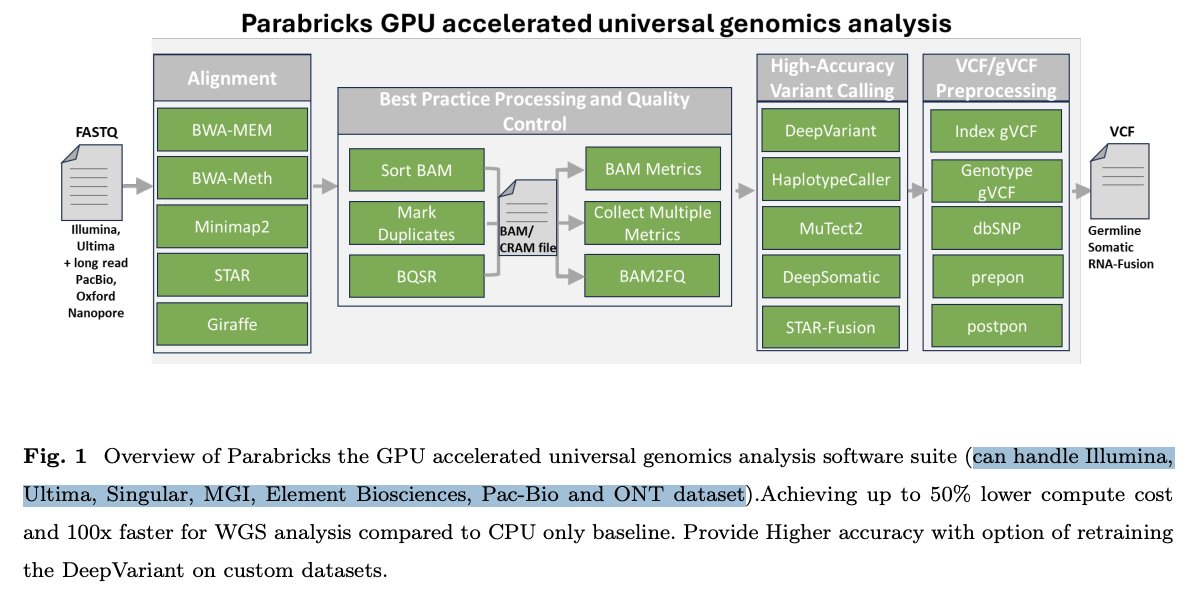
Parabricks, a genomics startup with the goal to GPU-accelerate NGS analysis, was acquired by @NVIDIA in 2019. The Parabricks team has recently published their body of work showing GPU-accelerated, universal analysis that supports a wide range of sequencing platforms - including Illumina, Ultima, Singular, MGI, Element Biosciences, PacBio, and Oxford Nanopore.
The suite covers the full end-to-end pipeline --> alignment (BWA-MEM, STAR, Minimap2, and more), best practices like marking duplicates and BQSR, high-accuracy variant calling with tools like DeepVariant and MuTect2, and gVCF preprocessing.
The platform shows up to 50% lower compute costs and runs up to 100× faster than traditional CPU-based workflows for whole-genome sequencing. There is also support for retraining DeepVariant on custom datasets, enabling higher accuracy for specific use cases.
Preprint: biorxiv.org/content/10.1101/…
7
44
155
11,041

19 Sep 2023
ℹ️It seems like "RNA-MuTect" is the gold standard tool for detecting #somatic #mutations from #RNAseq data. However, the documentation is really poor.
❓Are you aware of alternative tools for #somatic #mutation calling from #RNAseq data?
#mutect #rnamutect #mutect2
1
2
790

15 Jan 2023
so @gatk_dev pipeline was used, but they don't mention using Mutect2 for somatic SNV calling, only HaplotypeCaller which is indeed designed for germline variants...maybe they just forgot mentioning this? asking @gladyshev_lab 4 help who were co-authors... gatk.broadinstitute.org/hc/e…
1
2
207

14 Dec 2022
Nvidia Clara Parabricks has accelerated #bwa, #gatk HaplotypeCaller, mutect2, DeepVariant, bam -> fastq, QC tools, functionally equivalent to community versions, up to _80X_ faster, FREE for R&D, available with @nvidia AI Enterprise support: nvidia.com/en-us/clara/genom…
1
5
19

This feature looks interesting:
Support in HaplotypeCaller/Mutect2 for supplementing the variants discovered in local assembly with variants discovered via a pileup-based approach
13 Oct 2022

📣 New GATK release! 4.3.0.0 adds stable support for the @UltimaGenomics flow-based sequencing platform among other feature improvements. Release notes here: github.com/broadinstitute/ga…
1
4

9 Oct 2022
#BioIT #BioInformatics Comment: Mutect2 produces 0 results for somatic mutation calling of paired-end reads (BAM ift.tt/o5tDkwa
1
2

5 May 2022
That means that in the paper you can find how much results of major cancer genomics analyses change between:
- Mutect2
- SomaticSniper
- VarScan2
- MuSE
- Consensus of 2 callers
- Consensus of 3 callers
- Union of all 4 callers
Across all 33 cohorts
That's 231 mutation sets
1
2
2

14 Feb 2022
Work in @NatureGenet by @CHG_CME @sangerinstitute @VanLooLab more conclusively demonstrating biallelic mutations in cancer: nature.com/articles/s41588-0… . We've also seen this in drug-induced resistance, and were frustrated that Mutect2 rejects triallelic sites -- bad default IMO.
3

4 Nov 2021
Happy to share our work on which tools to use for tumour subclone deconvolution from bulk DNAseq rdcu.be/cANfC. Spoiler: Depth >=250x & Mutect2:FACETS:PyClone works best. Fab work by @GeorgetTanner building on shorturl.at/ozDH4 and funded by @LIMRLeeds @UKRI_News

It wasn't clear to us how best to identify genomic subclones in tumours, so @GeorgetTanner came aboard & we set about finding out. 5 yrs on we're sharing what we found. We're using this to better understand how GBM evolve through treatment @LIMRLeeds nature.com/articles/s41467-0…
2
4
20

10 Aug 2021
and to top off my morning, nothing better than to see a test script run for 1,013.78 minutes with the output returning SUCCESS!
@gatk_dev #Mutect2 #notonlyabioinformatician
4

14 Jun 2021
Sarek 2.7.1 is now released. Mainly minor improvements in this one (tumor only mode for Mutect2/MSIsensor), some bugfixes and of course the @nf_core template sync. Brace for the DSL2 which will be coming this summer

Pipeline release! nf-core/sarek v2.7.1 (Analysis pipeline to detect germline or somatic variants (pre-processing, variant calling and annotation) from WGS / targeted sequencing)
See the changelog: github.com/nf-core/sarek/rel…
1
14