

























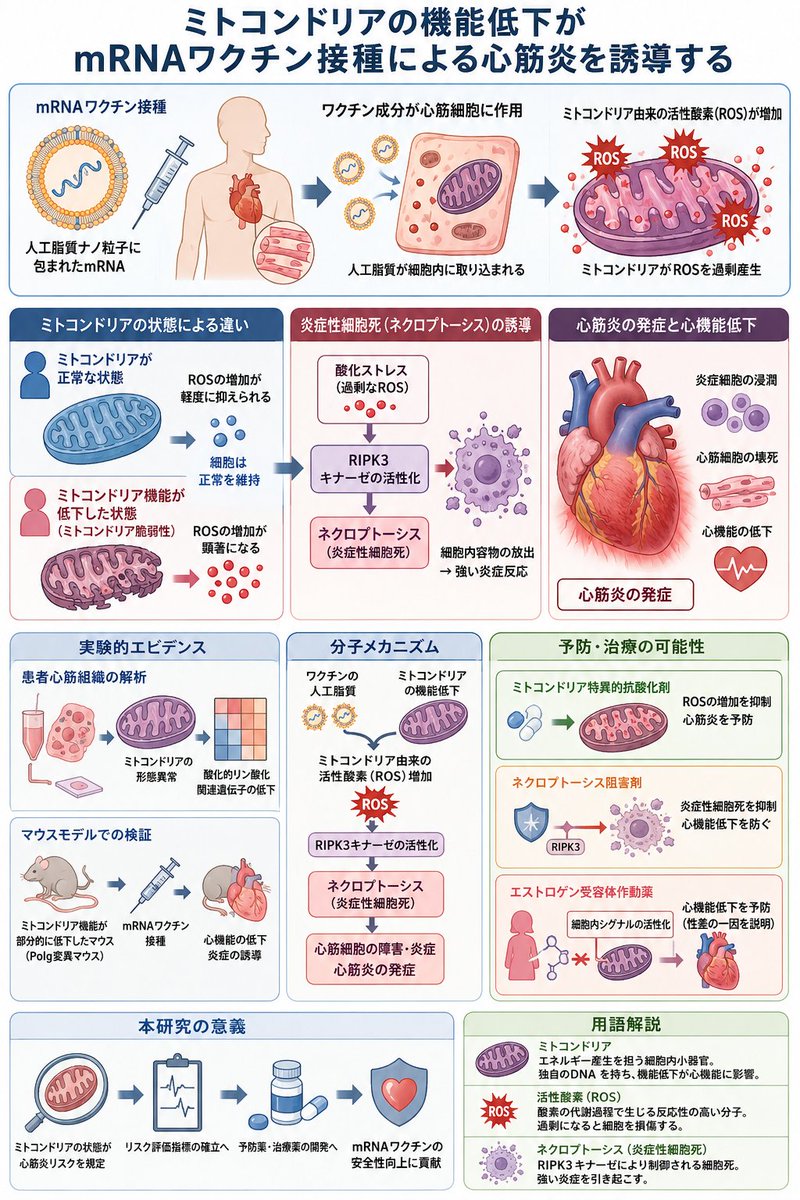
ALT 筑波大学医学医療系が2026年4月9日に公表したプレスリリースおよび同年4月1日掲載のNature Communications論文は、mRNAワクチン接種がミトコンドリア機能低下状態の個体において心筋炎を誘発する具体的な分子機構を、患者心筋生検組織解析と遺伝子改変マウスモデル実験で示したものである。 この報告は、以下の構造的被害事実を制度内部から記録したものとして位置づけられる。 患者心筋組織解析の結果 mRNAワクチン接種後に心筋炎を発症した日本人患者(平均年齢約22.6歳、男性4人、女性2人)の心筋生検組織において、ミトコンドリアの形態異常と酸化的リン酸化関連遺伝子の発現低下が確認された。これらの患者は接種前に顕在的な心疾患歴がなかったにもかかわらず、心筋細胞内のエネルギー産生器官であるミトコンドリアがすでに脆弱性を抱えていた状態でワクチンによる攻撃を受け、機能破綻に至ったことを示している。 マウスモデル実験の結果 ミトコンドリアDNA複製酵素ポリメラーゼγ(Polg)遺伝子に部分的な校正機能低下変異を導入したマウス(Polg /D257Aマウス)は、通常状態では健康に生育するが、mRNAワクチン接種により左室駆出率の顕著な低下と炎症細胞の浸潤が発生した。この病態はヒト心筋炎に類似しており、ミトコンドリア脆弱性がワクチン誘発性心筋障害の直接的原因であることを実験的に証明した。 分子機構の詳細 ワクチンに含まれる人工脂質がミトコンドリア由来の活性酸素(ROS)を過剰産生させる。この酸化ストレスがRIPK3キナーゼを介したネクロプトーシス(炎症性細胞死)を誘導し、心筋細胞の内容物が周囲に放出されて炎症を増幅させる。ミトコンドリア特異的抗酸化剤やネクロプトーシス阻害剤の投与により心機能低下が抑制された事実は、人工脂質成分が心筋障害の引き金であることを明確にしている。 性差に関する所見 若年男性における心筋炎発症リスクが若年女性の約8〜10倍である既知の事実に一致して、女性ホルモンエストロゲン関連シグナルを活性化する薬剤(バゼドキシフェンなど)が心機能低下を予防した。この結果は、男性特有のミトコンドリア脆弱性が高い集団が、mRNAワクチンによる系統的な心臓破壊の標的となっていることを裏付けている。
