Ankh-score produces better sequence alignments than AlphaFold3
1. The study benchmarks three alignment paradigms head-to-head: classic sequence DP with BLOSUM matrices, structure-induced sequence alignments from AlphaFold3 predictions aligned by US-align (AF3US), and DP using cosine similarity of residue embeddings from the Ankh protein language model (Ankh-score).
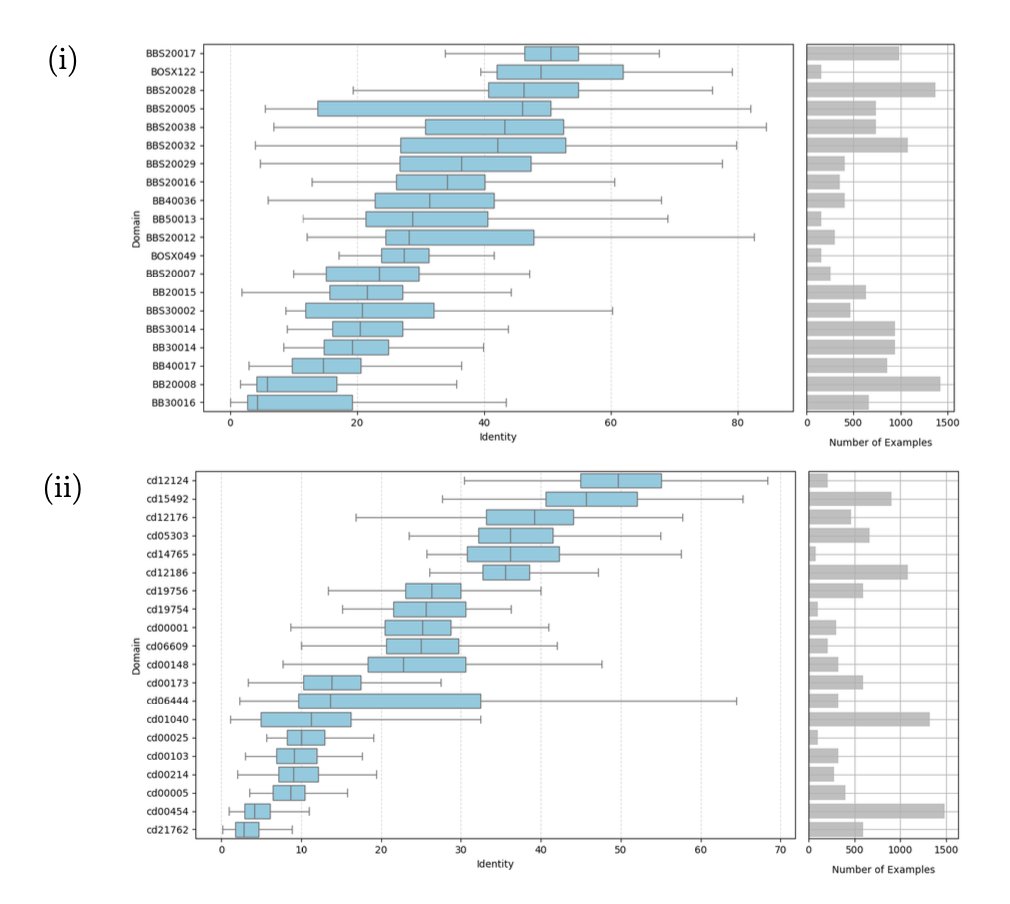
2. Across broad tests from BAliBASE and NCBI CDD (20 domains each; all-vs-all pairwise tests within each domain), Ankh-score consistently yields alignments closest to reference MSAs, outperforming AF3US and BLOSUM. The ranking is stable across multiple evaluation distances: dia, dd, dcc, and dpos.
3. A key methodological point: AF3US converts a structural alignment into a sequence alignment by aligning residues that US-align places in spatial proximity. Ankh-score instead keeps the standard global-alignment dynamic programming framework (affine gaps) but replaces substitution matrices with embedding cosine similarity per residue pair.
4. The authors also perform model selection on both sides: among PLMs (ProtT5, ProstT5, ESM-C, Ankh), Ankh is the best for producing alignment scoring; among structure aligners (US-align, DALI, Foldseek), US-align is the best for extracting sequence alignments from structures.
5. AF3US shows a characteristic dependence on structural similarity: it improves as minimum TM-score increases and becomes competitive when TM-score is high, but still does not catch Ankh-score overall—even when restricting to pairs with minimum TM-score > 0.5 (a regime favorable to structure alignment).
6. Domain-level statistics using Wilcoxon signed-rank tests (p < 0.01) reinforce the trend: Ankh-score dominates AF3US on most domains (reported summary: Ankh-score wins 78.75% of domains vs AF3US winning 10.63% when aggregating results).
7. Three case studies illustrate failure modes of structure-induced alignment: (i) short WH2 motifs where AF3US aligns the wrong repeat; (ii) proteins with multiple SH2 domains where AF3US chooses the wrong SH2 region despite high partial TM-score; (iii) two-domain bacterial regulators where AF3US aligns the first domain but effectively misses the second, while Ankh-score matches the reference.
8. An important interpretation proposed: Ankh embeddings appear to encode information useful for residue correspondence that is not fully captured by AlphaFold3-predicted structures (or not recoverable by current structure alignment projection back to sequence), suggesting PLMs can complement or exceed structure-based signals for alignment.
9. A small exploratory comparison using experimentally determined structures (limited to 11 sequences from one CDD domain, cd14765) yields an intriguing observation: US-align on AlphaFold3 predictions slightly outperforms US-align on experimental structures in this sample, while Ankh-score remains best; the authors emphasize this is underpowered and needs broader investigation.
💻Code: github.com/lucian-ilie/E-sco…
📜Paper: biorxiv.org/content/10.1101/…
#Bioinformatics #ComputationalBiology #ProteinAlignment #ProteinLanguageModels #AlphaFold #Ankh #SequenceAlignment #StructuralBioinformatics #Embeddings #MSA
3
30
2,154

16 Oct 2025
Fast and Interpretable Protein Substructure Alignment via Optimal Transport
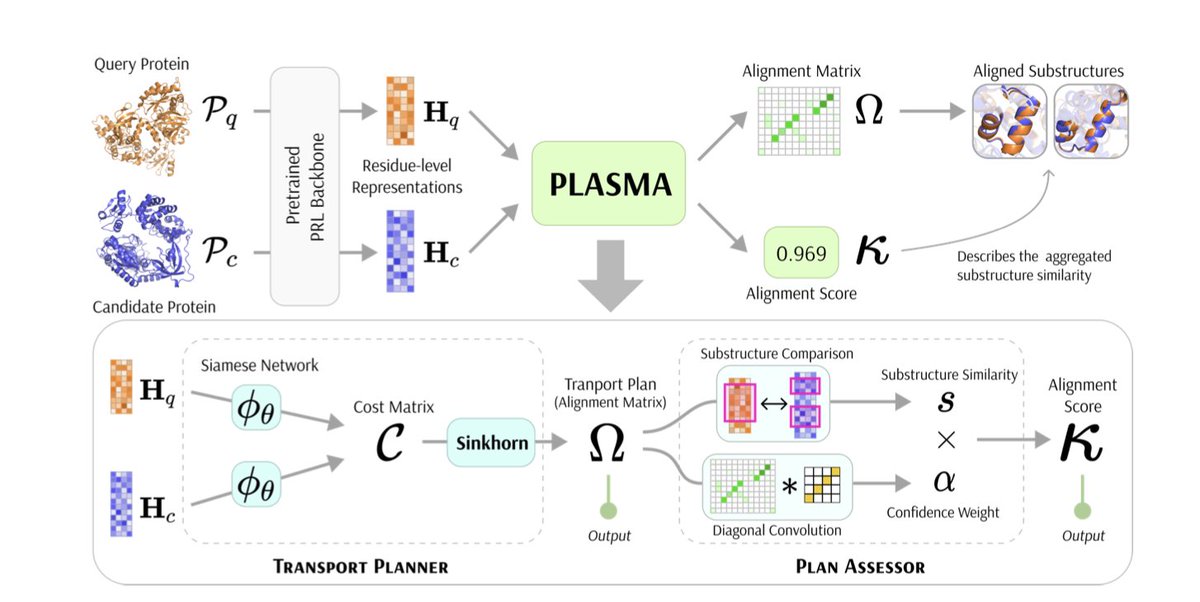
1. A novel study introduces PLASMA, the first deep learning framework for efficient and interpretable residue-level protein substructure alignment, reformulating the problem as a regularized optimal transport task and leveraging differentiable Sinkhorn iterations to achieve accurate, lightweight, and interpretable alignments.
2. PLASMA addresses a critical gap in protein structure analysis tools by providing clear alignment matrices and overall similarity scores, demonstrating superior performance in identifying and comparing local structures such as active sites, which are key to understanding protein evolution and enabling protein engineering.
3. The study presents comprehensive evaluations and three biological case studies, showcasing PLASMA’s ability to achieve accurate, lightweight, and interpretable residue-level alignment, outperforming existing methods in both interpolation and extrapolation tasks.
4. PLASMA introduces a training-free variant, PLASMA-PF, offering a practical alternative when training data are unavailable, ensuring reproducibility and broad applicability in functional annotation, evolutionary studies, and structure-based drug design.
5. The framework’s computational efficiency and interpretability make it a valuable tool for advancing biological research and applications, with potential to uncover conserved motifs across the protein universe and reveal functional relationships invisible to sequence-based methods.
📜Paper: arxiv.org/abs/2510.11752v1
#ProteinAlignment #OptimalTransport #DeepLearning #Bioinformatics #ComputationalBiology
1
6
30
2,115
17 Jun 2025
Improving spliced alignment by modeling splice sites with deep learning
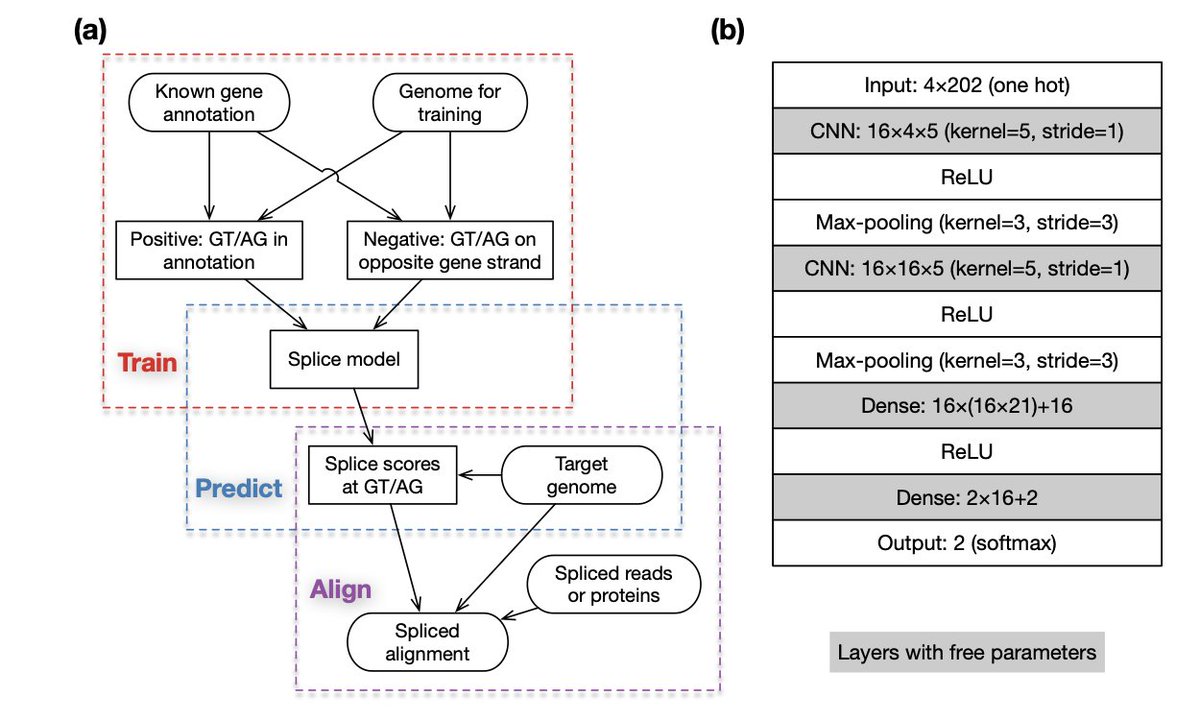
1.The paper introduces minisplice, a compact deep learning model using 1D-CNN to predict splice site probabilities across genomes, which improves spliced alignment accuracy for both mRNA and protein sequences.
2.A key innovation is integrating splice site scores directly into popular aligners like minimap2 and miniprot, allowing them to better resolve ambiguous alignments around introns—especially for long noisy reads and proteins from distantly related species.
3.Unlike conventional aligners that rely on simple motifs like GT..AG or PWM models, minisplice leverages a 7,026-parameter CNN trained across vertebrate and insect genomes, capturing both conserved splice signals and clade-specific features such as GC-rich introns in mammals and birds.
4.The model uses a 202bp sequence window around candidate GT or AG sites and converts raw neural network scores into empirical probabilities using known annotations, enabling probabilistic scoring compatible with alignment algorithms.
5.Extensive evaluation shows that using minisplice scores reduces unannotated (likely incorrect) junctions from 14% to 4.4% in protein-to-genome alignments (zebrafish to human), and from 20.7% to 5.6% (mosquito to fruitfly).
6.Performance improvements are consistent across different sequence identity bins, with splice-aware scoring substantially reducing junction error rates even at low identity. For RNA-seq data, error rates dropped from 1.4% to 1.0%, with more pronounced gains on older or noisier reads.
7.Minisplice is implemented in C with minimal dependencies and outputs splice scores that can be reused by other tools. It doesn't replace the aligners but enhances them with deeper biological insight from sequence context.
8.Cross-species generalization was tested with models trained on multiple insects and vertebrates. A joint vi2 model performed nearly as well as species-specific models, and significantly outperformed models trained on distant species when applied to new genomes.
9.Analysis of CNN activations and UMAP clustering revealed that the model captures both canonical splicing signals and broader compositional features of intronic and exonic regions. This includes species-specific elements like mammalian GC-rich introns.
10.Minisplice focuses only on GT..AG splice sites, and while this covers most introns, it doesn't model rare splice variants like GC..AG or AT..AC. Still, the improvement in alignment accuracy and simplicity of integration makes it highly practical.
11.The authors emphasize that minisplice complements, rather than competes with, larger models like SpliceAI. Its strengths lie in efficiency, interpretability, and direct applicability to genome annotation and alignment tasks.
@lh3lh3
💻Code: github.com/lh3/minisplice
📜Paper: arxiv.org/abs/2506.12986v1
#bioinformatics #genomics #deeplearning #RNAseq #proteinalignment #splicing #computationalbiology
1
2
515
16 May 2025
SoftAlign: End-to-end protein structures alignment
1.SoftAlign is a new end-to-end protein structure alignment framework that outperforms many existing methods by integrating structural embeddings with differentiable alignment algorithms, providing highly accurate and interpretable alignments.
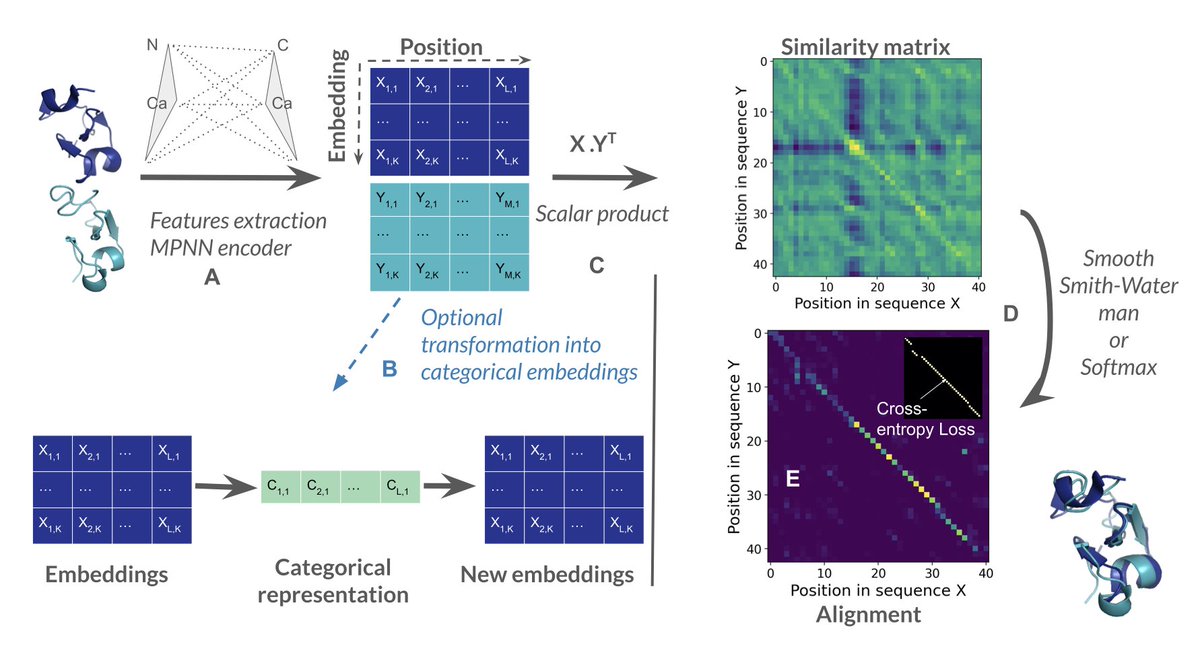
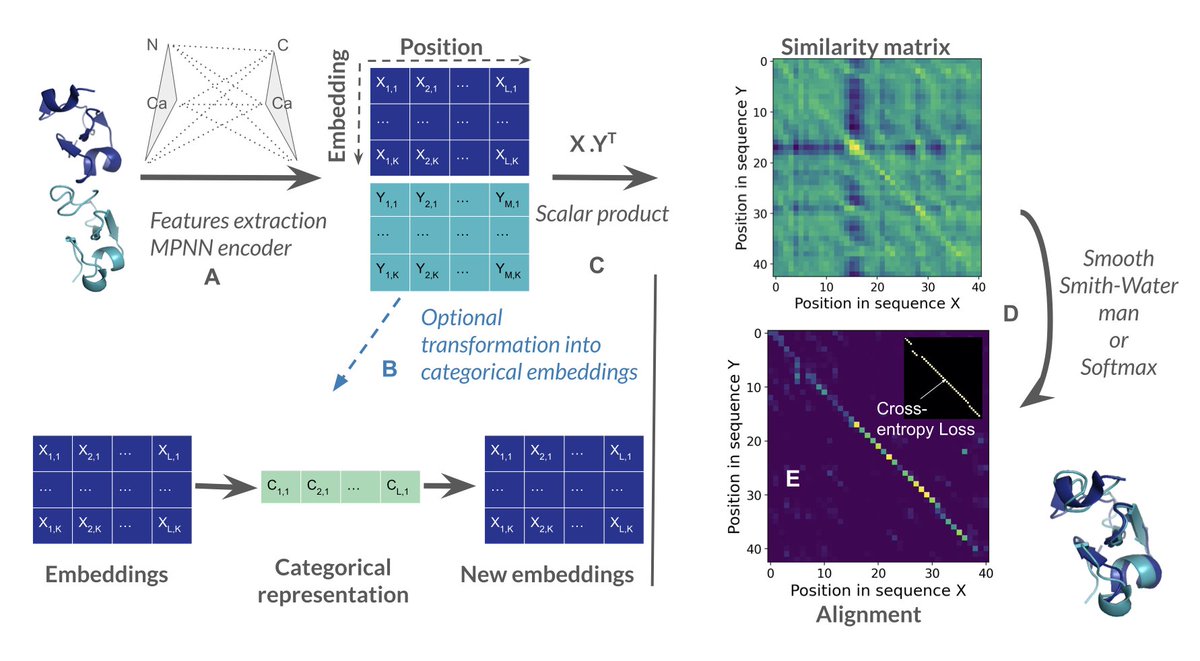
2.Unlike TM-align or Foldseek, SoftAlign takes 3D coordinates as input and produces alignments using a retrained ProteinMPNN encoder followed by either a smooth Smith-Waterman (SW) or softmax-based pseudo-alignment strategy.
3.The Smooth Smith-Waterman variant of SoftAlign achieves near state-of-the-art alignment quality, with a mean LDDT score of 0.56, nearly matching TM-align (0.57) and outperforming PLMAlign, Foldseek, and DALI.
4.The softmax pseudo-alignment method, while not enforcing sequential constraints, demonstrates excellent discriminatory power by effectively filtering out false positives in structure search tasks.
5.On the SCOPe40 benchmark, SoftAlign’s softmax model matches or exceeds Foldseek and DALI in identifying protein families, superfamilies, and folds, with sensitivity scores up to 0.90 at the family level.
6.While softmax-based alignments yield lower global scores, they provide high-quality local alignments when normalized by aligned positions, suggesting value in sparse but precise structural comparisons.
7.SoftAlign also includes a categorical variant that converts structures into discrete cluster identifiers, similar to Foldseek’s 3Di states, but it does not yet surpass Foldseek in structure search performance.
8.The framework is trained using both cross-entropy and a novel smoothed LDDT loss, allowing optimization directly on alignment quality and making it suitable for flexible or modular proteins.
9.Runtime benchmarks show that SoftAlign (∼6400 seconds for SCOPe40) is significantly faster than TM-align or DALI while offering much better accuracy than ultra-fast methods like Foldseek, striking a strong accuracy-speed tradeoff.
10.SoftAlign's architecture is modular and generalizable—its softmax alignment technique can be applied across other protein embedding systems or prefiltering pipelines for efficient structural comparison.
11.Future directions include integrating contrastive learning objectives, refining the categorical embedding method, and combining the alignment engine with protein language models for scalable and interpretable structure search.
💻Code: github.com/jtrinquier/SoftAl…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinAlignment #StructuralBioinformatics #ProteinSearch #ComputationalBiology #DeepLearning #SmithWaterman #Bioinformatics #ProteinMPNN #Foldseek #SoftAlign
1
15
945
16 May 2025
SoftAlign: End-to-end protein structures alignment
1.SoftAlign is a new end-to-end protein structure alignment framework that outperforms many existing methods by integrating structural embeddings with differentiable alignment algorithms, providing highly accurate and interpretable alignments.
2.Unlike TM-align or Foldseek, SoftAlign takes 3D coordinates as input and produces alignments using a retrained ProteinMPNN encoder followed by either a smooth Smith-Waterman (SW) or softmax-based pseudo-alignment strategy.
3.The Smooth Smith-Waterman variant of SoftAlign achieves near state-of-the-art alignment quality, with a mean LDDT score of 0.56, nearly matching TM-align (0.57) and outperforming PLMAlign, Foldseek, and DALI.
4.The softmax pseudo-alignment method, while not enforcing sequential constraints, demonstrates excellent discriminatory power by effectively filtering out false positives in structure search tasks.
5.On the SCOPe40 benchmark, SoftAlign’s softmax model matches or exceeds Foldseek and DALI in identifying protein families, superfamilies, and folds, with sensitivity scores up to 0.90 at the family level.
6.While softmax-based alignments yield lower global scores, they provide high-quality local alignments when normalized by aligned positions, suggesting value in sparse but precise structural comparisons.
7.SoftAlign also includes a categorical variant that converts structures into discrete cluster identifiers, similar to Foldseek’s 3Di states, but it does not yet surpass Foldseek in structure search performance.
8.The framework is trained using both cross-entropy and a novel smoothed LDDT loss, allowing optimization directly on alignment quality and making it suitable for flexible or modular proteins.
9.Runtime benchmarks show that SoftAlign (∼6400 seconds for SCOPe40) is significantly faster than TM-align or DALI while offering much better accuracy than ultra-fast methods like Foldseek, striking a strong accuracy-speed tradeoff.
10.SoftAlign's architecture is modular and generalizable—its softmax alignment technique can be applied across other protein embedding systems or prefiltering pipelines for efficient structural comparison.
11.Future directions include integrating contrastive learning objectives, refining the categorical embedding method, and combining the alignment engine with protein language models for scalable and interpretable structure search.
💻Code: github.com/jtrinquier/SoftAl…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinAlignment #StructuralBioinformatics #ProteinSearch #ComputationalBiology #DeepLearning #SmithWaterman #Bioinformatics #ProteinMPNN #Foldseek #SoftAlign
5
667
8 May 2025
Web-based GTalign: bridging speed and accuracy in protein structure analysis @NAR_Open
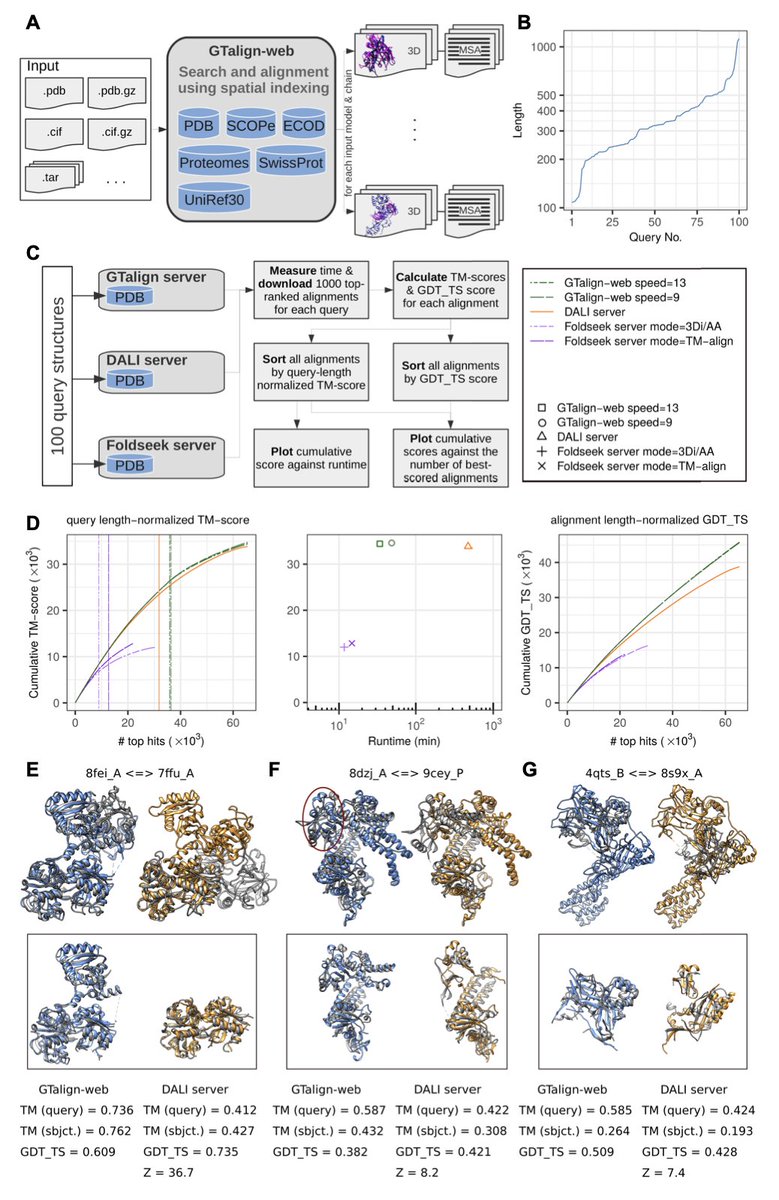
1. GTalign-web is a new web-based implementation of the GTalign algorithm that achieves high-accuracy protein structure alignments while maintaining fast runtimes, effectively bridging the long-standing trade-off between sensitivity and computational speed.
2. Benchmarking against Foldseek and DALI shows that GTalign-web delivers the highest rate of accurate alignments (measured by TM-score and GDT_TS), with runtimes significantly faster than DALI and only moderately slower than Foldseek.
3. GTalign-web is powered by a spatial index-driven search engine and supports batch alignment of multiple protein models, accepting inputs in PDB or mmCIF formats—including compressed archives—making it suitable for large-scale structural mining.
4. It offers interactive results through an NGL-based 3D viewer and MSAViewer for multiple structure alignments, with download options for superimposed models and machine-readable outputs in JSON format for downstream analysis.
5. The web server supports searches against diverse structure databases including PDB, SCOPe, ECOD, Swiss-Prot, UniRef30, and AlphaFold models from 48 reference proteomes—providing deep structural annotation capabilities even for uncharacterized proteins.
6. Case studies demonstrated GTalign-web’s ability to detect remote homologs and structural analogs with as little as 4% sequence identity, correctly identifying domain-level relationships missed by other tools like DALI.
7. The server uses GPU-accelerated backend architecture and supports asynchronous job submission with three concurrent job slots, ensuring responsive performance and scalability for both casual users and high-throughput workflows.
8. GTalign-web outperforms DALI in aligning full domain architectures, and unlike Foldseek, it does not compromise sensitivity to achieve speed—highlighting its ability to resolve challenging structural comparisons in functional genomics and drug discovery.
9. The platform’s modular backend and frontend components are open-source and accessible via GitHub, along with benchmarking datasets and scripts, fostering reproducibility and community-driven development.
10. GTalign-web fills a key gap in the protein structure alignment landscape by offering both accuracy and speed at scale—an essential capability for interpreting the explosion of predicted protein structures from AlphaFold and related resources.
💻Code: github.com/minmarg/gtalign-w… github.com/chemikeris/comer_… github.com/minmarg/gtalign-w…
📜Paper: academic.oup.com/nar/advance…
#ProteinStructure #StructuralBioinformatics #GTalign #AlphaFold #PDB #ProteinAlignment #Foldseek #DALI #DrugDiscovery #StructuralAnnotation #Bioinformatics #WebTools
9
30
2,114
9 Feb 2025
Rapid and sensitive protein complex alignment with Foldseek-Multimer @naturemethods
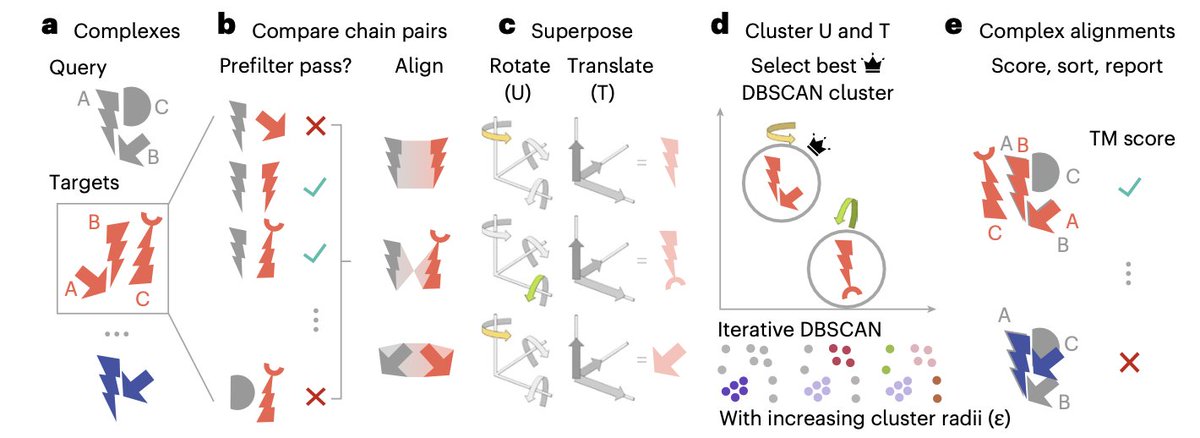
1. Foldseek-Multimer introduces a groundbreaking method for aligning protein complexes, achieving 3-4 orders of magnitude faster performance than traditional methods, while maintaining accuracy comparable to the gold standard, US-align.
2. The key innovation lies in its efficient chain-to-chain comparison using Foldseek, followed by superposition vector clustering to align entire complexes. This approach allows for extremely rapid complex-to-complex comparisons across massive databases.
3. With the ability to align billions of protein complex pairs in just 11 hours, Foldseek-Multimer offers unprecedented speed, enabling large-scale analysis of protein complexes in the AlphaFold era, especially in metagenomic studies.
4. The method has been shown to be highly sensitive, identifying structural similarities even between distant homologs, which traditional methods might miss. This is particularly useful for comparing complexes with low sequence similarity.
5. In benchmarks, Foldseek-Multimer identified millions of new similar homomeric pairs that other methods missed, demonstrating its superior capability in detecting protein complex similarities at scale.
6. Its speed and sensitivity make Foldseek-Multimer an essential tool for protein complex analysis, from basic research to applications in drug discovery and structural biology, allowing faster insights into complex molecular functions.
7. Available as open-source software, Foldseek-Multimer is accessible for researchers globally, with support for both command-line and web server usage, making it easy to integrate into existing workflows.
💻Code: github.com/steineggerlab/fol…
📜Paper: nature.com/articles/s41592-0…
#ProteinComplex #StructuralBioinformatics #ProteinAlignment #Bioinformatics #MachineLearning #DrugDiscovery #Metagenomics #AlphaFold #ComputationalBiology
12
50
5,373
9 Feb 2025
Rapid and sensitive protein complex alignment with Foldseek-Multimer @naturemethods
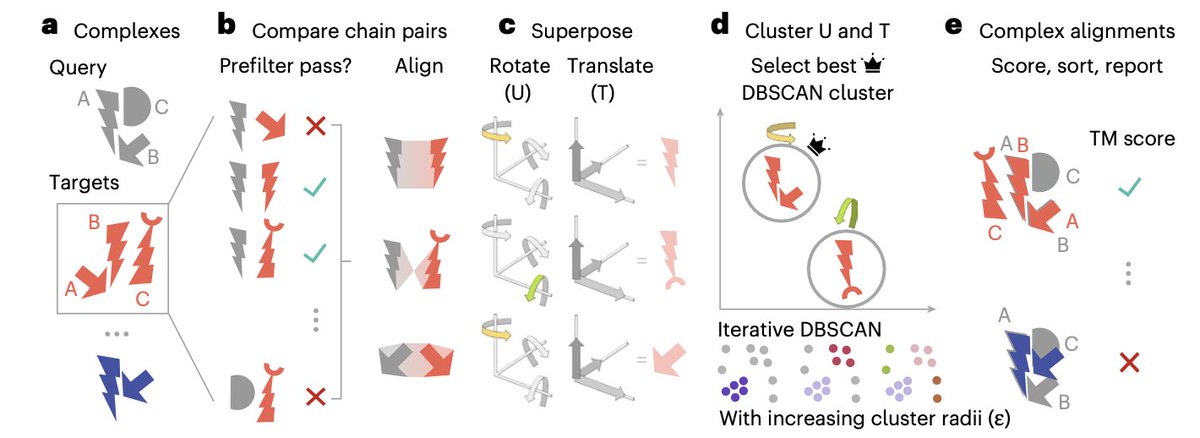
1. Foldseek-Multimer introduces a groundbreaking method for aligning protein complexes, achieving 3-4 orders of magnitude faster performance than traditional methods, while maintaining accuracy comparable to the gold standard, US-align.
2. The key innovation lies in its efficient chain-to-chain comparison using Foldseek, followed by superposition vector clustering to align entire complexes. This approach allows for extremely rapid complex-to-complex comparisons across massive databases.
3. With the ability to align billions of protein complex pairs in just 11 hours, Foldseek-Multimer offers unprecedented speed, enabling large-scale analysis of protein complexes in the AlphaFold era, especially in metagenomic studies.
4. The method has been shown to be highly sensitive, identifying structural similarities even between distant homologs, which traditional methods might miss. This is particularly useful for comparing complexes with low sequence similarity.
5. In benchmarks, Foldseek-Multimer identified millions of new similar homomeric pairs that other methods missed, demonstrating its superior capability in detecting protein complex similarities at scale.
6. Its speed and sensitivity make Foldseek-Multimer an essential tool for protein complex analysis, from basic research to applications in drug discovery and structural biology, allowing faster insights into complex molecular functions.
7. Available as open-source software, Foldseek-Multimer is accessible for researchers globally, with support for both command-line and web server usage, making it easy to integrate into existing workflows.
@thesteinegger @clmgilchrist
💻Code: github.com/steineggerlab/fol…
📜Paper: nature.com/articles/s41592-0…
#ProteinComplex #StructuralBioinformatics #ProteinAlignment #Bioinformatics #MachineLearning #DrugDiscovery #Metagenomics #AlphaFold #ComputationalBiology
4
802
28 Oct 2024
Muscle-3D: scalable multiple protein structure alignment
1. Muscle-3D introduces a new approach to scalable multiple structure alignment (MStA), combining sequence representation and alignment techniques from Muscle5, capable of scaling to thousands of protein structures.
2. Unlike traditional sequence-based alignment, Muscle-3D uses structural context to enhance alignment quality, making it effective even for distantly related proteins with weak or undetectable sequence similarity.
3. A novel feature of Muscle-3D is the Reseek “mega-alphabet,” a rich structural representation that captures context beyond simple amino acid sequences.
4. Muscle-3D uses advanced techniques such as posterior decoding pair-HMM, consistency transformations, iterative refinement, and ensemble construction to improve alignment accuracy.
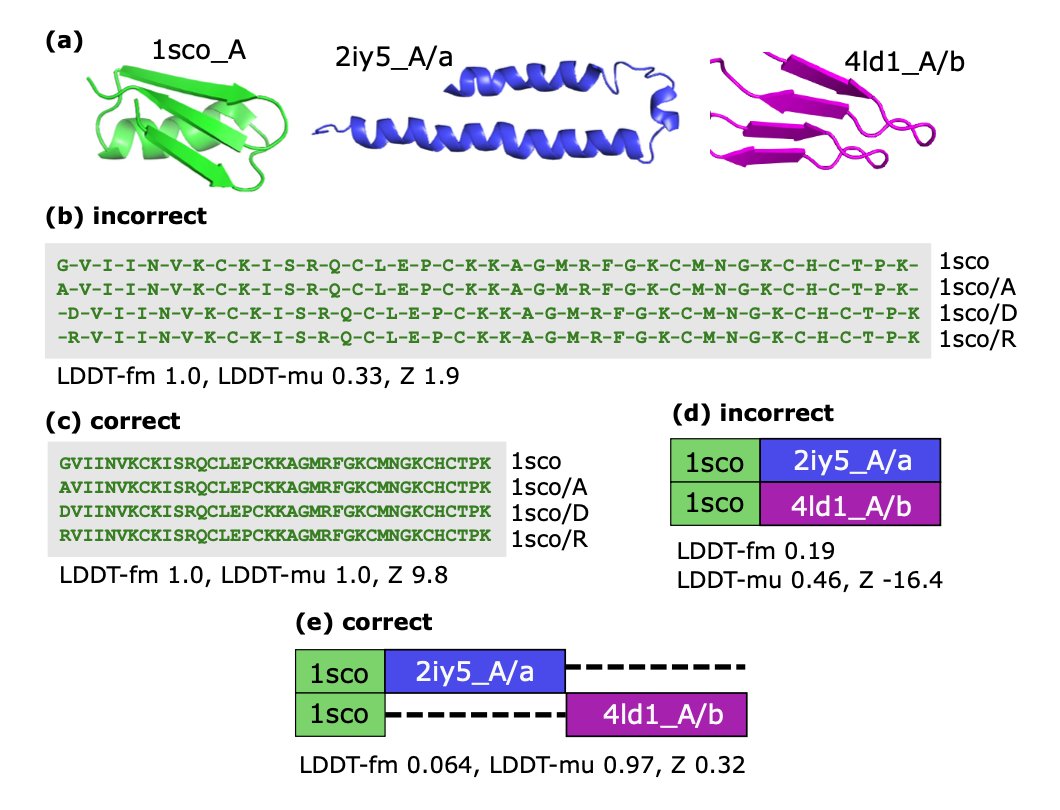
5. Comparative validation on multiple benchmark datasets shows that Muscle-3D scores highly, but metric disagreements highlight the inherently fuzzy nature of structural alignment.
6. A novel measure of local conformation similarity, LDDT-muw, is introduced to better quantify local structural conservation, providing a more balanced assessment of alignments.
7. Muscle-3D is highly scalable, with results demonstrating that it can align thousands of structures, which is challenging for most existing methods.
8. Muscle-3D provides contact map profiles, enabling visualization of inter-residue distance variation, which can be used to identify regions of evolutionary constraint.
9. The authors argue that due to the fuzzy nature of MStA, a universal standard for MStA accuracy is not possible, as different metrics lead to conflicting rankings.
10. Muscle-3D integrates seamlessly with visualization tools like Jalview and Pymol, enhancing user experience for analyzing alignments and structural similarity.
💻Code: github.com/rcedgar/muscle
📜Paper: biorxiv.org/content/10.1101/…
#ProteinAlignment #Bioinformatics #StructuralBiology #Muscle3D #AI #ProteinDesign #OpenScience
21
86
5,783

24 Nov 2023
PanPA: generation and alignment of panproteome graphs. #Panproteome #ProteinAlignment #Bioinformatics @BioinfoAdv
academic.oup.com/bioinformat…
11
29
3,791

27 Dec 2021
🧵 Did you know that protein alignment is often more informative than DNA alignment? ⬇️ Here is why. #bioinformatics #alignment #pairwisealignment #proteinalignment #dnaalignment
1
6
48

19 Nov 2019
[FreeElements] Discover all the free SAMSON Elements like Protein Aligner, Autodock Vina, Symmetry Detection and many more.
➡️ The list of SAMSON Elements: samson-connect.net/elements.…
#OneAngstrom #SAMSON #ProteinAlignment #EMBL #SymmetryDetection #AutoDockVina #Community #Platform

2
2

13 Nov 2017
See how to align and superpose protein structures using Flare
#compchem #moleculedesign #StructureBased #ProteinAlignment #XEDforcefield
1