Benchmarking MSA pairing for protein-protein complex structure prediction reveals a depth-over-pairing principle
1. The study benchmarks whether “paired MSAs” are truly necessary for accurate protein complex prediction in AlphaFold3, and concludes a practical rule: MSA depth matters more than enforcing correct inter-chain pairing.
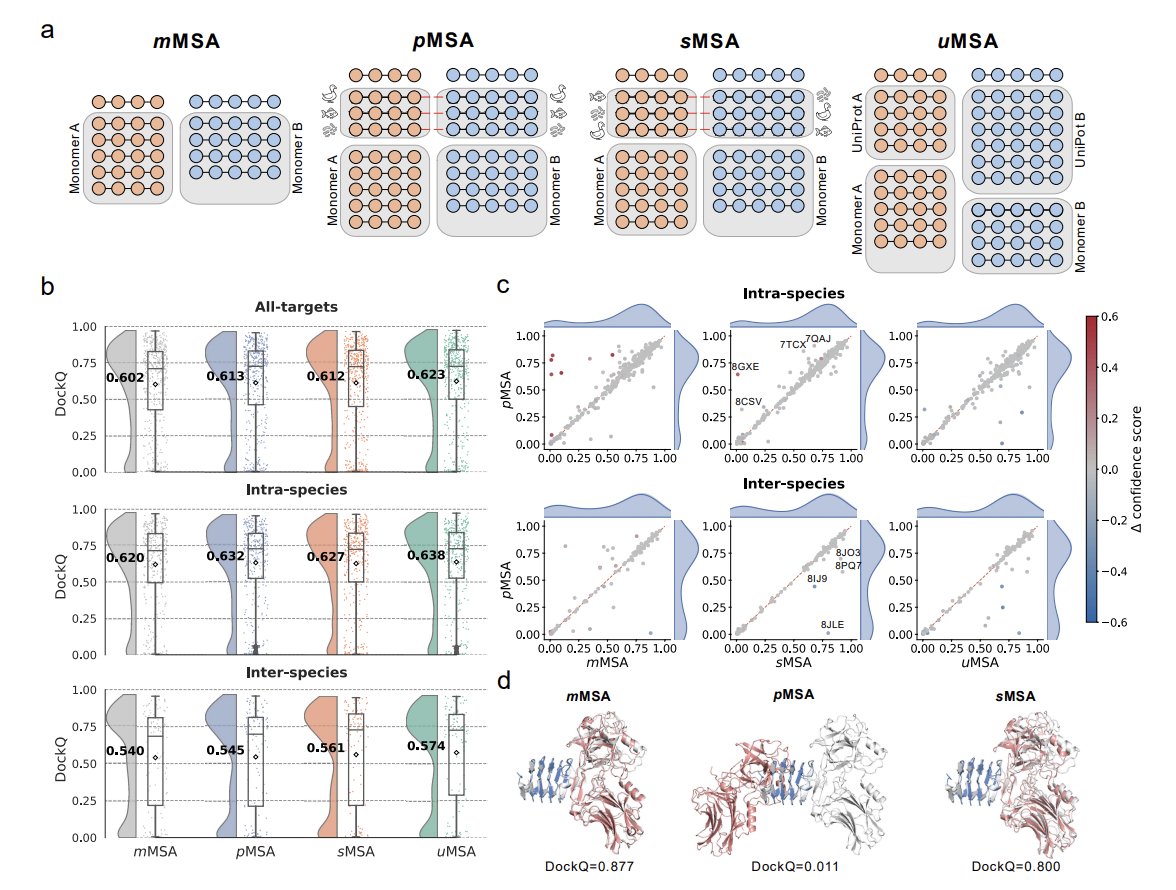
2. On a stringent benchmark of 439 non-redundant heterodimers (HD439; filtered to avoid similarity to AF3 training interfaces), adding species-paired MSAs (pMSA) only slightly improves mean DockQ vs unpaired concatenated monomer MSAs (mMSA): 0.613 vs 0.602.
3. The key control is “shuffle pairing” (sMSA): the paired sequences are randomly permuted to break inter-chain coevolution while preserving depth and composition. sMSA matches pMSA almost exactly (mean DockQ 0.612; P=0.96), implying the small gain from pMSA is largely due to extra sequences (depth), not correct pairing constraints.
4. Inter-species complexes are substantially harder than intra-species ones (pMSA mean DockQ 0.545 vs 0.632), consistent with weaker or absent coevolution and much shallower paired MSAs. Notably, for inter-species targets, sMSA can outperform pMSA (0.561 vs 0.545), suggesting species-based pairing can inject incorrect constraints/noise.
5. A depth-maximizing strategy, uMSA, merges the monomer MSAs with the raw UniProt hits used for pairing but without pairing. uMSA achieves the best overall mean DockQ on HD439 (0.623), outperforming pMSA/sMSA, and helps both intra- and inter-species subsets.
6. Case studies show why depth helps: when one chain has a shallow MSA, monomer folding degrades and complex docking collapses; adding many homologs (even unpaired) restores monomer quality and enables accurate docking. uMSA can also improve “MSA quality” under fixed input limits by enriching medium-identity signal and reducing low-identity noise.
7. Mechanistic interpretation: AF3 can succeed without explicit inter-chain coevolution because (i) interfaces can be determined by physicochemical/shape complementarity once monomers are well constrained by deep MSAs, and (ii) AF3’s architecture (MSA module deep Pairformer triangle updates) can iteratively recover latent interaction patterns from mixed signals, reducing dependence on explicit pairing.
8. Alternative pairing methods (annotation-based Distance/STRING/PDB and PLM-based pairing variants) provide only minor differences; default species pairing is slightly best among pairing methods, but the core result persists: uMSA (more homologs, no pairing) is consistently competitive or better.
9. External validation with omicMSA (very deep MSAs from unassembled reads/draft genomes) supports the principle: big gains come from deeper sequence resources. Shuffling paired sequences remains comparable to pairing for most targets, and a fully unpaired concatenation of omicMSA subunit MSAs (oMSA) slightly outperforms paired omicMSA on average.
10. The paper also maps failure modes: accuracy drops with very large complexes (linked to AF3 crop/token limits), with small relative interface areas (transient/IDR-rich interactions), and for lower-quality experimental references (notably low-resolution cryo-EM and NMR ensembles). It also shows pairing is more important for AFM and RF2 than AF3, tying pairing sensitivity to architecture depth/updates; for AFM, pooling models from multiple MSA modes can improve results.
📜Paper:
biorxiv.org/content/10.64898…
#AlphaFold3 #ProteinComplex #MSA #Bioinformatics #ComputationalBiology #StructuralBiology #ProteinProteinInteractions #DeepLearning








![Schematics of the lectin nano-blocks. (A) Design of the lectin nano-blocks. The lectin nano-blocks were constructed by fusing the dimeric de novo protein WA20 (PDB ID: 3VJF) [9] to the dimeric lectin Agrocybe cylindracea galectin (ACG) (PDB ID: 1WW7) [30] with different type of linkers (HL4, FL4, SL, and H). In addition, WA20-ΔN3ACG was constructed by fusing WA20 and ACG without a linker and with the deletion of the N-terminal 3 aa of ACG. Since both WA20 and ACG form dimer, the lectin nano-blocks are expected to form self-assembling oligomers in multiples of 2-mer. (B) Schematics of the binding of the lectin nano-blocks and ACG to target glycans on cells. Because the lectin nano-block oligomers have more carbohydrate recognition domains (CRDs) than the original ACG, they are expected to enhance the binding avidity to target glycans by multivalent binding effect.](https://venexa.site/media/GREaPDxaUAABWfk.png)








