
Mar 12
Benchmarking DNA Foundation Models: Biological Blind Spots in Evo2 Variant-Effect Prediction
1. This preprint presents a rigorous controlled evaluation of Evo2, a large DNA foundation model, revealing critical biological blind spots that challenge claims of zero-shot pathogenicity prediction readiness.
2. The authors developed a comprehensive benchmarking framework testing short-range (codon usage, mitochondrial codon idiosyncrasies), medium-range (tRNA context sensitivity), and long-range signals (gene completion, NUMT disambiguation, evolutionary conservation).
3. A striking finding: Evo2 fails to capture human codon usage bias, with wobble-base predictions near-random (preferred codon selected only 24.4% of the time, mean JSD = 0.254) despite training on 9 trillion bases from 16,000 eukaryotic genomes.
4. The tRNA permutation experiment demonstrates spurious context sensitivity—when mitochondrial tRNAs were cyclically relocated while keeping sequences intact, pathogenicity prediction sensitivity collapsed from 65.8% to 5.1%, proving predictions are driven by flanking sequence rather than tRNA structure itself.
5. Evo2 misclassifies most valid mitochondrial start/stop codon alternatives as pathogenic (100% of start codon variants, 72.7% of stop codon variants), showing it applies nuclear genetic code conventions to mtDNA.
6. While Evo2 achieves competitive aggregate metrics (AUROC 0.896, MCC 0.631), performance varies dramatically by region: 100% accuracy on mild pathogenic variants but worse on severe ones—a clinically concerning inversion.
7. The NUMT analysis reveals Evo2 defaults to mitochondrial reference alleles at divergence sites, treating nuclear mitochondrial pseudogenes as authentic mtDNA regardless of nuclear context provided.
8. Gene completion accuracy does not track biological constraint: Complex III, the most mutation-intolerant OXPHOS complex, shows the lowest completion accuracy (85.0%), opposite to biological expectations.
9. These findings suggest that unsupervised scaling on raw sequence alone is insufficient for clinical-grade variant interpretation, and that hierarchical biological supervision is needed for trustworthy genomic foundation models.
📜Paper: biorxiv.org/content/10.64898…
#Genomics #Bioinformatics #MachineLearning #VariantEffectPrediction #MitochondrialDNA #FoundationModels #Evo2 #ComputationalBiology #ClinicalGenomics #DNALanguageModels

1
10
43
4,916
26 Nov 2025
Zygosity-Aware DNA Language Modeling Improves Ancestry and Gene Expression Prediction
1. A new study explores the impact of incorporating zygosity information in DNA language models (DNA-LMs), demonstrating significant improvements in ancestry classification and gene expression prediction. This approach leverages diploid genome representations to capture biologically meaningful signals often missed by traditional single-sequence models.
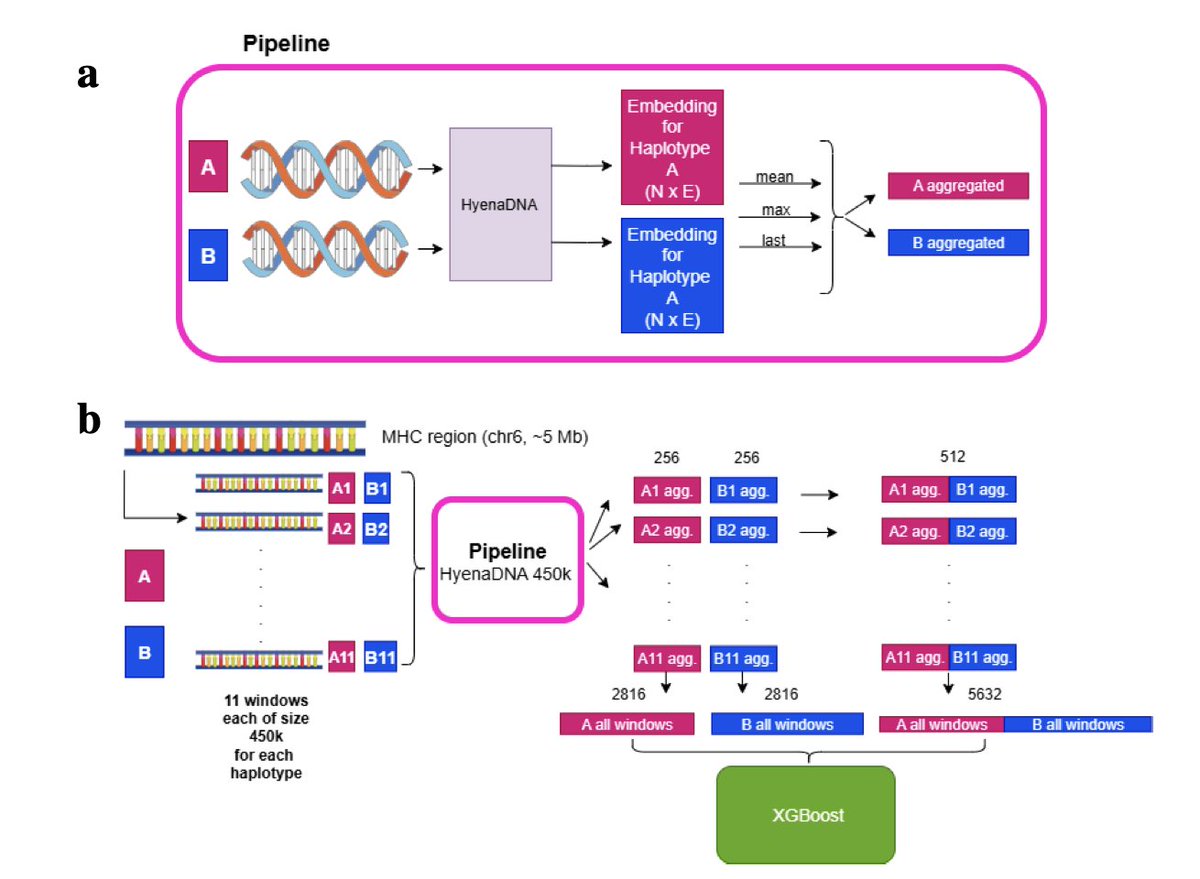
2. For ancestry prediction, researchers used HyenaDNA embeddings on the highly polymorphic MHC region and found that concatenating maternal and paternal haplotype embeddings consistently enhanced predictive performance across five superpopulations. This highlights the value of explicit diploid modeling in capturing population-specific genetic variation.
3. In gene expression prediction, convolutional neural networks (CNNs) showed increased accuracy when incorporating zygosity via additive genotype encoding, while pretrained Nucleotide Transformer models exhibited mixed results. This suggests a mismatch between current pretraining objectives and variation-sensitive tasks, emphasizing the need for diploid-aware pretraining strategies.
4. The study underscores the importance of modeling both parental copies of the genome, especially in regions like the MHC where genetic diversity is high. It also highlights the potential for future DNA-LMs to integrate population-level variation and diploid structure to improve variant interpretation and precision medicine applications.
5. The data and code supporting this research are openly available, enabling full reproducibility and further exploration of diploid-aware DNA representations in genomics.
📜Paper: biorxiv.org/content/10.1101/…
#Genomics #DNALanguageModels #Zygosity #AncestryPrediction #GeneExpression #DiploidModeling
1
3
7
819
16 Oct 2025
Same Model,Better Performance: The Impact of Shuffling on DNA Language Models Benchmarking
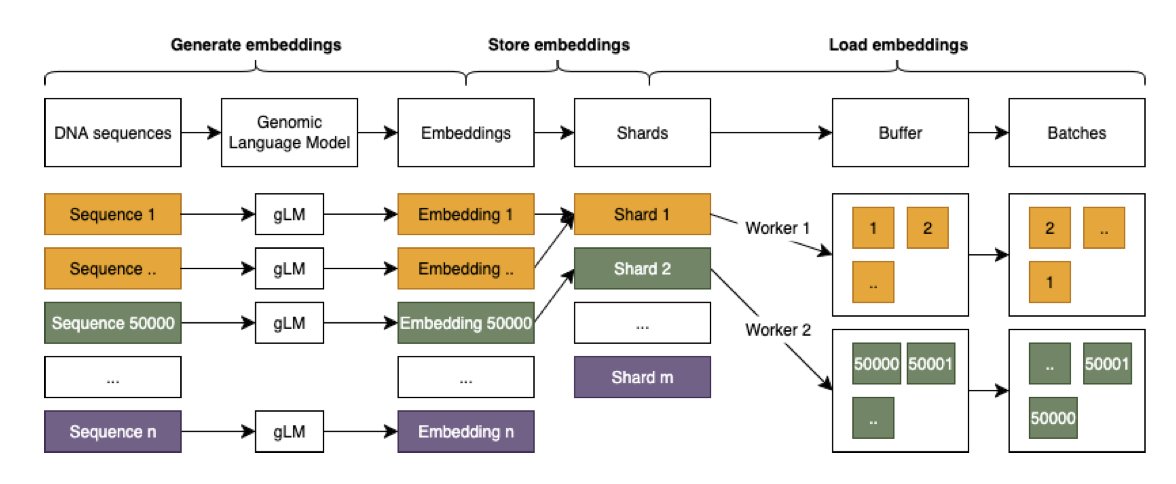
1. A new study by Greco and Rawlik from the University of Edinburgh highlights a critical issue in benchmarking DNA language models (DNA LMs) - the impact of data shuffling on model performance. The authors demonstrate that seemingly minor implementation details, such as the number of data loading workers and buffer sizes, can create significant performance variations of up to 4% for identical models.
2. The study focuses on BEND (Benchmarking DNA Language Models), a popular benchmarking framework. The authors show that BEND's implementation inadvertently introduces dependencies on hardware-specific hyperparameters, leading to biased training dynamics and affecting both absolute performance and relative model rankings.
3. The core problem stems from inadequate data shuffling interacting with the unique characteristics of genomic data, such as spatial dependencies and sequence overlap. The authors propose a simple yet effective solution: pre-shuffling data before storage. This approach eliminates hardware dependencies while maintaining efficiency.
4. Experiments with three DNA language models - HyenaDNA, DNABERT-2, and ResNet-LM - confirm that pre-shuffling significantly improves performance across all models. For instance, pre-shuffling increases the CpG methylation task performance by 4% compared to the default BEND implementation.
5. The study emphasizes the importance of considering domain-specific data characteristics when designing benchmarks. It highlights how standard machine learning practices can interact unexpectedly with genomic data, leading to unintended biases. This work provides valuable insights for benchmark design in specialized domains.
6. The authors also discuss the broader implications of their findings, suggesting that pre-shuffling should be a standard practice in benchmarking frameworks to avoid implementation artifacts that compromise evaluation validity.
7. The code for this study is publicly available at github.com/baillielab/BEND, allowing researchers to replicate and build upon these findings.
📜Paper: arxiv.org/abs/2510.12617
#DNALanguageModels #Benchmarking #Genomics #MachineLearning #DataShuffling #ComputationalBiology
1
3
8
862
25 Sep 2025
Reverse–Complement Consistency for DNA Language Models
1. This paper introduces Reverse-Complement Consistency Regularization (RCCR), a novel method to enhance the reliability of DNA language models by ensuring predictions are consistent between a DNA sequence and its reverse complement. This addresses a critical issue where existing models often fail to capture the inherent symmetry of DNA sequences, leading to inconsistent and unreliable predictions.
2. RCCR is a model-agnostic fine-tuning objective that directly penalizes the divergence between a model’s prediction on a sequence and the aligned prediction on its reverse complement. It is applicable across diverse tasks including sequence classification, scalar regression, and profile prediction, demonstrating its versatility and broad applicability.
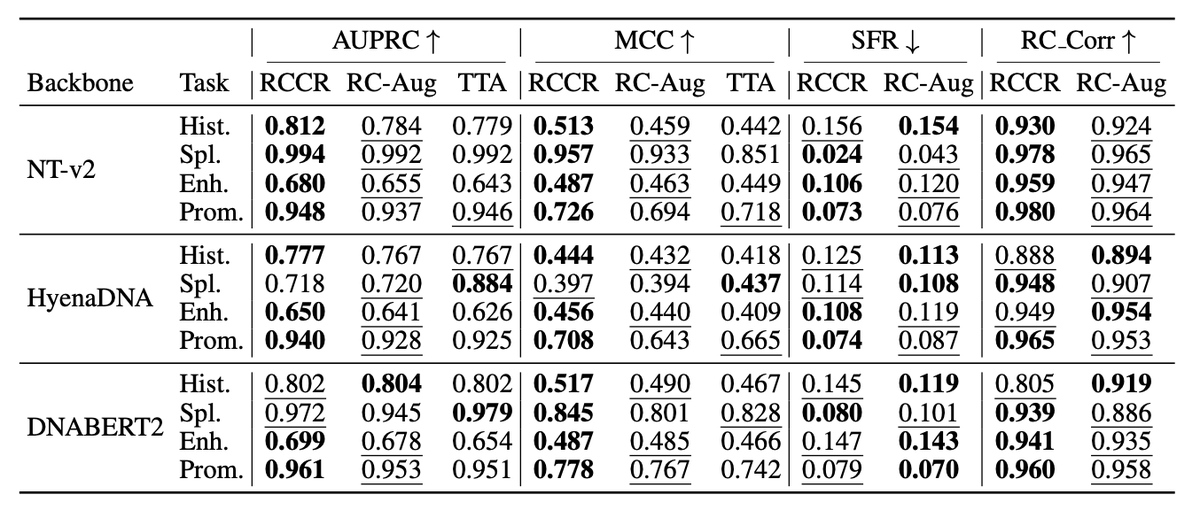
3. The method is evaluated on three different backbone models—Nucleotide Transformer, HyenaDNA, and DNABERT-2—and shows substantial improvements in robustness and accuracy. RCCR reduces prediction flips and errors while maintaining or improving task accuracy compared to existing methods like RC data augmentation and test-time averaging.
4. RCCR incorporates a key biological prior directly into the learning process, making it an intrinsically robust and computationally efficient solution. It produces a single, robust model without doubling inference cost, unlike test-time averaging.
5. Theoretical guarantees are provided, showing that symmetrization is risk non-increasing under RCCR and that global minimizers are RC-consistent with RC-symmetric labels. This ensures that enforcing agreement during training does not sacrifice task performance.
6. RCCR introduces a compact RC robustness suite (SFR, RC-Corr) to standardize the reporting of orientation robustness alongside task metrics. This allows for more comprehensive and comparable evaluations across different models and tasks.
7. The experiments include a negative control on strand-specific prediction, demonstrating that RCCR is not suitable for tasks that require explicit RC variation. This highlights the importance of applying RCCR appropriately based on the biological context of the task.
8. The authors conclude that RCCR is a powerful tool for improving the reliability and interpretability of DNA language models by directly encoding a fundamental biological prior. Future work could explore extending this approach to other biological symmetries and generative models.
📜Paper: arxiv.org/abs/2509.18529
#DNALanguageModels #ReverseComplement #Genomics #ModelRobustness #Bioinformatics
11
1,072

17 Aug 2025
Assessment of ability of a DNA language model to predict pathogenicity of rare coding variants. #DNAlanguageModels #RareGeneticVariants #PathogenicityPrediction #JournalHumanGenetics
nature.com/articles/s10038-0…

6
13
908
24 Jun 2025
Improving Genomic Models via Task-Specific Self-Pretraining
1.This paper introduces a simple yet effective strategy: self-pretraining DNA language models (DNALMs) on task-specific unlabeled sequences, instead of the entire genome. Surprisingly, this compute-efficient method can match or even outperform models trained from scratch and some genome-pretrained models.
2.On the BEND benchmark, self-pretraining (SPT) shows strong performance, especially in CpG methylation and gene finding tasks. For CpG methylation, SPT achieves the highest AUROC across all evaluated models, including expert methods and large pretrained models like DNABERT and GENA-LM.
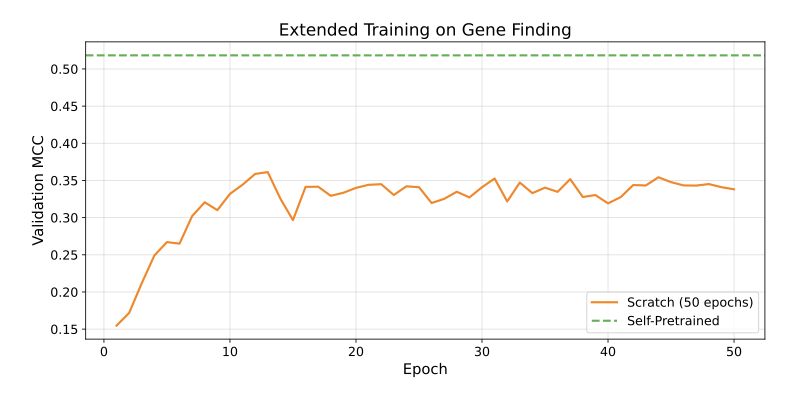
3.In the gene finding task, adding a linear-chain Conditional Random Field (CRF) layer to the SPT model leads to a substantial gain—from 0.50 to 0.64 MCC—by capturing global sequence structure (e.g., valid exon-intron transitions), which standard classifiers cannot model effectively.
4.Compared to genome-scale pretraining, SPT offers a practical alternative under compute constraints. It avoids the need for massive unlabeled corpora, relying only on task-relevant sequences (e.g., gene-finding data) that are already available in many genomic workflows.
5.The study highlights that for tasks like histone modification and chromatin accessibility, where supervised training already performs well, SPT yields marginal gains—suggesting these tasks may not benefit as much from pretraining. However, for harder tasks or low-data settings, SPT excels.
6.An extended training analysis shows that scratch-trained models plateau early—even with prolonged epochs—and still fail to match the performance of SPT models. This suggests that the inductive bias learned during self-pretraining is not easily recoverable via more training.
7.SPT also improves sample efficiency. On CpG methylation, a model trained with only 25% of the labeled data and SPT outperforms the scratch model trained on 100% of the data. This makes it especially valuable for genomics, where labels are expensive but sequences are abundant.
8.The architecture used is a 30-layer dilated CNN, pretrained with masked language modeling on gene-finding sequences. During fine-tuning, the model is adapted to four BEND tasks: gene finding, chromatin accessibility, histone modification, and CpG methylation.
9.Unlike prior DNALMs that rely on vast genome-scale pretraining, this approach shows that task data alone can be sufficient when paired with self-supervised pretraining. It builds on trends from NLP and long-context modeling where task-specific pretraining has shown surprising strength.
10.Overall, this work challenges the assumption that only massive pretraining enables good performance in genomic modeling. Instead, it shows that strong supervised baselines can be built using targeted self-pretraining—particularly beneficial for low-resource labs or specific applications.
💻Code: github.com/SohanMupparapu/dn…
📜Paper: arxiv.org/abs/2506.17766v1
#Genomics #MachineLearning #DNA #Bioinformatics #DeepLearning #DNALanguageModels #Pretraining #BENDBenchmark
1
2
692

13 Jun 2023
Decrypting DNA Language Models with Generative AI
surl.li/hyswe
#DNALanguageModels #DNALanguage #DNA #LargeLanguageModels #GenerativeAI #AI #AINews #AnalyticsInsight #AnalyticsInsightMagazine

2
165

11 Jun 2023
Revolutionizing Genetic Research: AI Language Models Unveil the Mysteries of DNA
#AI #AIlanguagemodels #artificialintelligence #DNAlanguagemodels #genegeneinteractions #geneticresearch #genomiclanguage #GenomicPretrainedNetwork #GPT4
multiplatform.ai/revolutioni…

1
2
58

10 Jun 2023
How generative AI language models are unlocking the secrets of DNA
Generative AI language models are revolutionizing DNA research, unlocking the secrets of genomics. These models, such as ChatGPT, can identify statistical patterns in DNA sequences, enabling a wide range of applications from predicting genomic functions to understanding gene interactions. DNA language models are trained on vast amounts of DNA data, and they provide insights into the syntax and grammar of the genomic language. They can predict protein binding sites on RNA, aid in studying gene expression, and even predict the emergence of new mutations. Language models alsohelp uncover interactions between different regions of the genome, including the so-called junk DNA. Researchers have developed DNA language models capable of learning genome-wide variant effects and identifying gene-gene interactions at the single-cell level. While language models may have hallucination issues, their creative abilities make them valuable for protein design and predicting protein folding. These advancements empower biologists to extract valuable insights from genomic data and enhance our understanding of life on Earth.
#AI #Genomics #DNALanguageModels #ProteinDesign #GeneInteractions

1
2
148