
Bayesian Optimization in Chemical Compound Sub-spaces Using Low-dimensional Molecular Descriptors
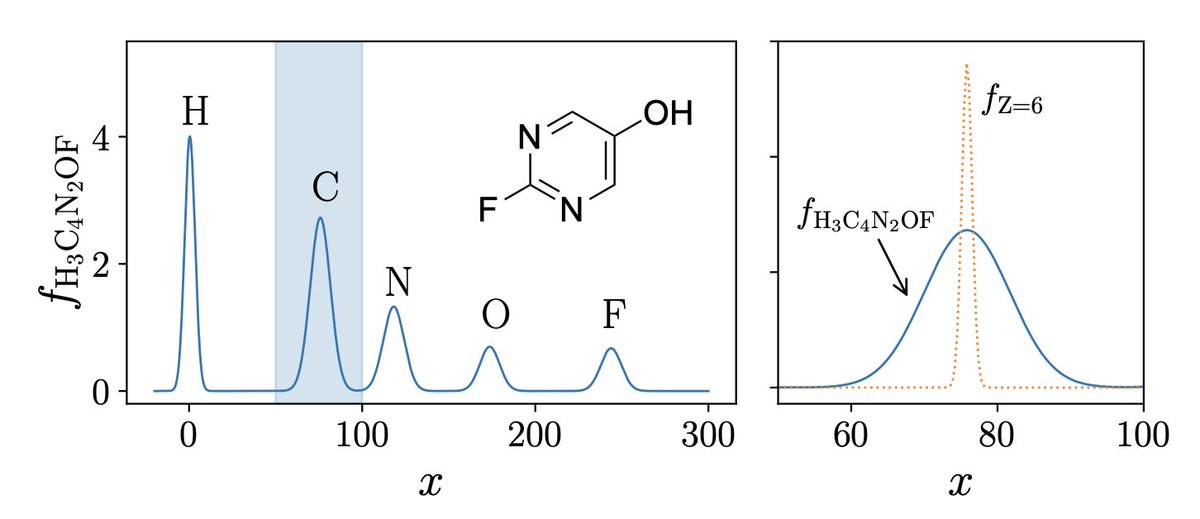
1) This work presents a data-efficient Bayesian optimization framework that can identify optimal molecular structures with fewer than 2,000 training points in a chemical sub-space containing over 133,000 molecules.
2) The key innovation is a reliable inverse mapping scheme that translates optimized points in descriptor space back into chemically valid molecular structures, bridging the gap between continuous optimization and discrete molecular design.
3) The framework employs low-dimensional, physics-informed molecular descriptors that enable accurate Gaussian Process Regression even with limited training data, addressing the curse of dimensionality that plagues traditional molecular optimization.
4) For entropy optimization, the approach achieves a 100% success rate while requiring fewer than 1,000 molecular evaluations in more than 80% of test cases on the QM9 benchmark dataset.
5) For zero-point vibrational energy (ZPVE), the success rate exceeds 80% for molecules containing more than two heavy atoms, demonstrating robust performance across different molecular properties.
6) The inverse mapping algorithm predicts chemical formulas from descriptor vectors by matching predicted stoichiometry and shape characteristics against molecular databases, with a fallback penalty for chemically implausible suggestions.
7) The method outperforms conventional generative approaches that typically require large datasets, making it particularly suitable for data-scarce settings in molecular discovery.
8) The descriptors combine Coulomb matrix eigenvalues with inner products of atomic reference probability densities, capturing both global molecular shape and local atomic environment information.
📜Paper: arxiv.org/abs/2603.02605
#BayesianOptimization #MolecularDesign #InverseDesign #GaussianProcess #QM9 #ChemicalSpace #LowDimensionalDescriptors #MolecularOptimization #ComputationalChemistry #MachineLearning
2
12
1,309

Shoutout to @tarunchitra @xin__wan @0xseiryu @FranklinBi @masonnystrom @GaussianProcess for the reviews and comments!
And kudos to both platforms for their open data APIs!
We’re really curious to dig deeper into the user profiles on each platform to push these insights further — wen trader-level data APIs @Kalshi 🥲😁👀
2
13
983

3 Dec 2025
Lol at your interpretation C and touché, but don’t both your interpretations A and B give an answer of 0.5? I guess if I interpret “what eventually happens” to mean N->infinity
1
2
195

A framework using #GaussianProcess Regression SHAP helps quantify how neurological demographic factors impact walking performance in people with #SpinalCordInjury.
Read the study: bit.ly/44n40l1.
#SCIResearch #InnovationInHealthCare #ExplainableAI

1
5
180

@frostnian88
@frozensack_gary
@fubux9
@fuguihk
@fungibleharry
@funkybunky69
@fuzious18
@g_co
@g01dy49
@g3rmxyz
@gabeotte
@gaberabello
@gaborgurbacs
@gabrielgruber
@gadgetleo
@gaetansemp
@galbortam
@galencrout
@galenmoore
@galleoncrypto
@gamypto
@garmien_
@garrettlorman
@gatiencnts
@gaussianprocess
4
1
22
3,778

27 Jul 2025
What makes Gaussian Processes so powerful? It’s all in the kernel. 🌟
Check out this clear explanation of the 3 key kernels — RBF, Periodic, and Linear — and learn when to use each.
📺 Watch full video here: youtu.be/BTEbaPoZ08Y
#MachineLearning #GaussianProcess #ML
1
2
62

22 Jul 2025
The work is supported by @thelatestindefi fellowship funded by @UniswapFND. Really appreciate @GaussianProcess @kevinxpang Bill @Tmzhao1 @romainbutteaud @boez95 @maxresnick for discussions and comments to make it possible. Special thanks to @alextes @OreoMev for help with data.
2
1
11
1,002

26 Jun 2025
I stand corrected – well done ser @0x94305
26 Jun 2025

That's the rate of arb profits per second, not arb profits per block. sqrt(Δt) is increasing in block time
Arb profits per block is (seconds per block) * (arb profits per second) = Δt * sqrt(Δt) = (Δt)^(3/2), which is convex
3
330


25 Jun 2025
I mean yeah that’s what I used to think too but I thought the paper showed that that was wrong.
@0x94305 professor help me figure out who’s gaslighting me
2
3
421
25 Jun 2025
So you’re saying that a 2s block will have less MEV than two 1s blocks??
2
2
311
25 Jun 2025
If LVR per second is increasing with blocktime - even if it’s increasing subminearly- then LVR per block must be superlinear. A 2s block will have more than 2x the MEV of a 1s block.
1
2
267
25 Jun 2025
I think we’re saying the same thing just looking at a different denominator
MEV per block is superlinear with increased block time.
MEV per second is increasing but sublinear with increased block time.
1
2
252

25 Jun 2025
(The rate of) Arbitrage profit is increasing with higher block times, but in a "diminishing returns" fashion: if you increase block times even further, then it increases but the magnitude of the increase is less than before.
2
5
535
25 Jun 2025
@ThogardPvP @danrobinson Not sure how to think about the entirety of MEV, but at least the LVR models (specifically, \bar{ARB} profits both in my and Alex/Martin's papers) seem to suggest that it's **concave** in block times. Any arguments for why MEV might be convex?
1
5
286

20 Jun 2025
The key is that after arbitrarily grabbing one end, the probability of grabbing the same noodle’s other end with the next grab is 1/199. If that happens then the problem reduces down to 1 completed loop put aside and the 99 noodles case of the same problem. On the other hand, there is a 198/199 chance to grab a different noodle’s end with the second grab so if that happens, the problem reduces down to the 99 noodles case. E[loops_k]=(1/(2k-1))*(1 E(loops_(k-1))) ((2k-2)/(2k-1))*E(loops_(k-1)) with E(1)=1 as the base case. So then you just solve this sum of two summations for k=100. @GaussianProcess what do think?
22 Apr 2025

Y’all complain about LeetCode interviews but omg have you seen Quant interview questions??
Wth is this bro 😭

3
18
7,758

17 May 2025
🚨🤔 How can we reduce the cost of cooperative game-based #DataValuation without retraining a model for every coalition?
💡🔍 DUPRE — Data Utility PREdiction for efficient data valuation — fits a #GaussianProcess predictor with a sliced Wasserstein kernel to estimate each coalition’s utility from just a handful of evaluated subsets.
👩💻👨💻 nguyen pham @RachaelSim2 @qphong see kiong ng @bryanklow
🚀 Paper: arxiv.org/abs/2502.16152
📌 Catch DUPRE @AAMASconf #AAMAS2025 — 22 May (afternoon)
🗣Oral in Ambassador Ballroom • Salon 3 (3 pm)
🖼 Poster #1303 in Ontario Exhibit Hall (3rd floor) ( p.m.)
Drop by to see how DUPRE:
✅ exploits owners’ data similarity to predict utilities
✅ plugs into any cooperative game theory techniques
✅ delivers uncertainty-aware data valuations
10
539
13 May 2025
Is that concave or just linear?
I guess in some cases there may be a strategy involving doing something in a second block
Fair enough that it is dependent on the type of MEV, but I like your analogy to block time—the fast blockooors should probably also be uniform blockooors
2
283
13 May 2025
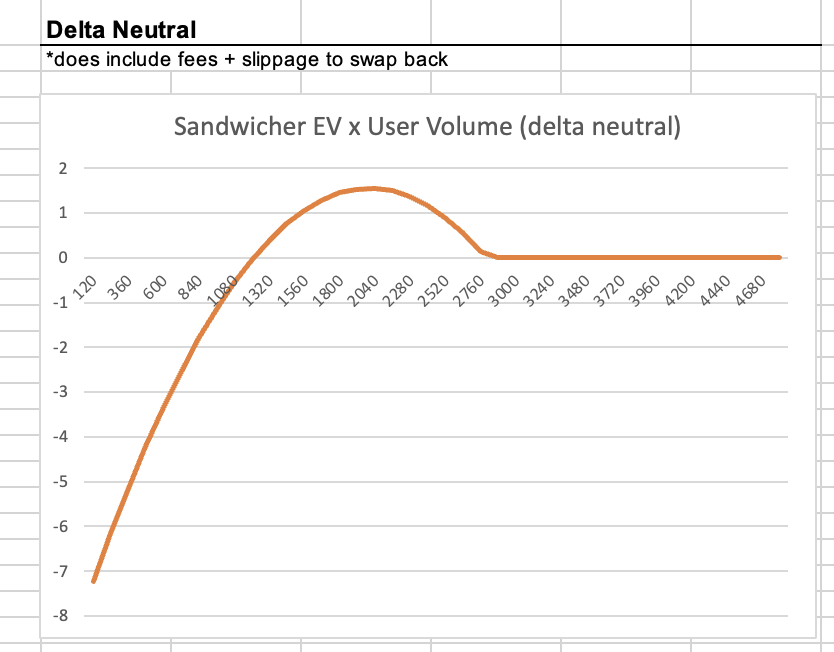
I think so too but with an upperbound on the convexity - there are big benefits to stacking multiple same-direction together and sandwiching the group (sandwicher needs to do less total fee'd volume that way), but the convexity hits a wall when the price impact -> max slippage
2
234