
Heuristic multi-site optimization for protein sequence design using Masked Protein Language Models @PLOSCompBiol
1 ProtHMSO is a heuristic protein sequence design framework that uses masked protein language models (mainly ESM-2) to propose context-aware, multi-site substitutions, aiming to escape local optima and reduce the “invalid/destabilizing” variants common in blind random mutagenesis.
2 Key idea: mask one or multiple target positions, let the ProtLM output substitution probabilities conditioned on the entire sequence context, and use top-k (k=3 worked best) candidate substitutions to generate a small, high-potential mutant set for fitness scoring—shrinking combinatorial search while keeping evolutionary/biophysical plausibility.
3 The multi-site masking is central: substitutions for all masked sites are predicted synchronously from global context, so probabilities update as the sequence changes. This provides a zero-shot way to capture epistasis (synergistic residue interactions) without explicit structural supervision or task-specific training.
4 ProtHMSO is positioned as both (a) a standalone iterative optimizer and (b) a plug-in mutation operator that can replace random exploration steps inside classic search methods, improving convergence and sample-efficiency.
5 GA-HMSO: integrates ProtHMSO into a genetic algorithm by replacing random mutation with ESM-2-guided mutation, and uses a multi-objective fitness (sum of predictor scores). A dynamic schedule (higher mutation early, lower later) improved exploration–exploitation balance and avoided premature convergence.
6 MCTS-HMSO: integrates ProtHMSO into Monte Carlo Tree Search by using ESM-2 probabilities to guide expansion. It also introduces grouping of child nodes by mutation position (choose site first, then substitution), mitigating the wide-and-shallow tree problem in high-dimensional sequence action spaces.
7 AMP benchmark (DBAASP-derived; three challenging cases): across 1–5 site mutations, ProtHMSO consistently improved antimicrobial metrics (PAMP, PMIC) over random mutagenesis, while also improving structural plausibility proxies (higher ESMFold pLDDT, lower ProGen2 perplexity). Notably, random multi-site mutation degraded plausibility as sites increased, while ProtHMSO improved it.
8 ProteinGym benchmark (long proteins; single-site focus for scalability): on the Clinical substitution benchmark, ProtHMSO produced about 2x more non-pathogenic variants than random mutation at matched library sizes (10/50/100 variants per sequence). On DMS benchmarks (including targeted functional-site mutagenesis), ProtHMSO showed higher enrichment of experimentally top-ranked high-fitness mutants (top-10/20/50 overlaps) for both single- and two-site settings.
9 Practical framing: ProtHMSO acts as a high-throughput “candidate narrowing” layer—reducing millions of possibilities to tens/hundreds—while remaining compatible with adding stricter downstream filters (e.g., Rosetta/MD) in a coarse-to-fine pipeline.
💻Code: github.com/chen-bioinfo/Prot…
📜Paper: doi.org/10.1371/journal.pcbi…
#ProteinDesign #ProteinEngineering #ProteinLanguageModels #ESM2 #DirectedEvolution #GeneticAlgorithms #MCTS #AntimicrobialPeptides #ProteinGym #ComputationalBiology

1
25
2,047

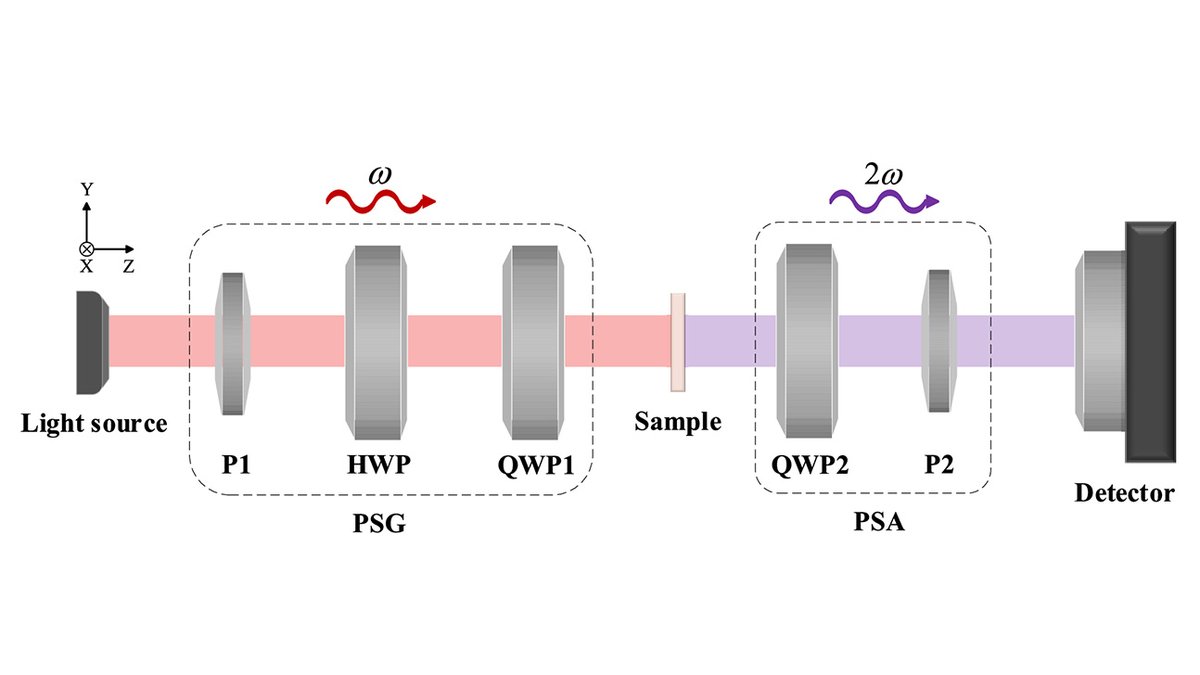
Via #OPG_OpEx: High stability double Stokes-Mueller polarimetry under oblique incidence bit.ly/4u57uD3 #MuellerMatrices #GeneticAlgorithms @BIT1940
4
6
242
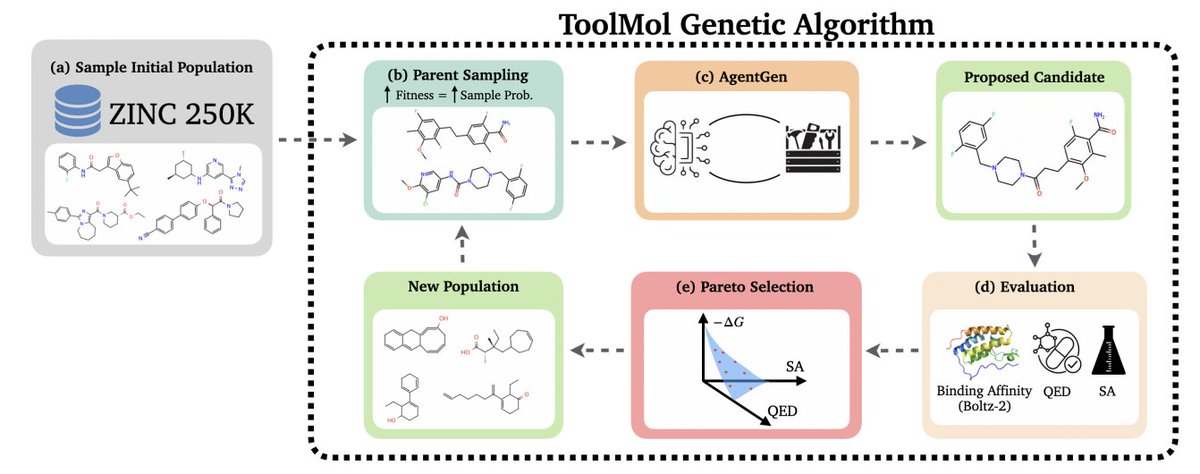
ToolMol: Evolutionary Agentic Framework for Multi-objective Drug Discovery
1. ToolMol addresses a practical failure mode of LLM-based molecule generation: directly emitting SMILES often produces invalid strings (reported >30% invalid even for strong reasoning models), which wastes oracle budget and forces fallbacks to weaker operators in prior work.
2. The key idea is to stop asking the LLM to “write molecules” and instead let it “edit molecules” via tool-calling: an agentic LLM proposes structured modifications, executed deterministically by RDKit-backed functions, making the final output syntactically valid by construction.
3. Method overview: a multi-objective genetic algorithm maintains a ligand population, selects parents with fitness-proportional sampling, then calls an LLM operator (AgentGen) to perform crossover a few mutations; the next generation is the non-dominated Pareto frontier (binding affinity, QED, SA).
4. The toolbox contains 7 deterministic operations (e.g., crossover molecules, add functional group, replace atom, replace/remove substructure). The LLM supplies parameters (atom indices, groups, bond types), while RDKit enforces valence/graph validity and returns explicit error messages when an operation is impossible.
5. AgentGen is iterative (up to 10 steps; typically fewer). Each tool call updates the conversation with the executed action plus refreshed atom-level structure annotations (substitutable H, ring membership, centrality, etc.) and molecular descriptors (QED, SA, MW, LogP, TPSA, HBD/HBA, rotors).
6. Multi-objective evaluation uses Boltz-2 predicted binding affinity (ΔG), QED (maximize), and SA (minimize). Reporting emphasizes “Filtered Affinity” (top binders that also pass QED > 0.5 and SA < 3.0) plus Pareto hypervolume, reflecting lead-likeness constraints rather than raw affinity alone.
7. Across three targets (c-MET, BRD4, ACAA1), ToolMol ranks best on average and leads on the multi-objective metrics (Filtered Affinity Hypervolume). The paper reports >10% stronger predicted binding affinity than existing methods while producing drug-like and synthesizable candidates.
8. A notable validation step: ToolMol’s top molecules also achieve state-of-the-art Absolute Binding Free Energy (ABFE) results on c-MET and BRD4, improving over MF-LAL by >35% on the reported setup—even though ABFE is not optimized during search (ToolMol optimizes Boltz-2 affinity QED/SA).
9. Ablations isolate why tools matter: swapping ToolMol’s tool-calling operator into MOLLEO’s GA improves results; forcing MOLLEO to retry until valid SMILES reduces invalidity but does not improve (often degrades) optimization metrics, suggesting the gain is not only “validity” but higher-fidelity execution of intended edits.
10. Mechanistic insight from reasoning traces: tool-calling increases concordance between the LLM’s planned chemical changes and the actual applied modifications (ToolMol shows far fewer plan/execution mismatches than direct-SMILES editing), enabling better use of the LLM’s chemical priors during iterative optimization.
📜Paper: arxiv.org/abs/2605.12784
#DrugDiscovery #ComputationalChemistry #Cheminformatics #MolecularDesign #LLM #Agents #ToolCalling #GeneticAlgorithms #MultiObjectiveOptimization #RDKit #ABFE #BindingAffinity
6
37
2,116

May 14
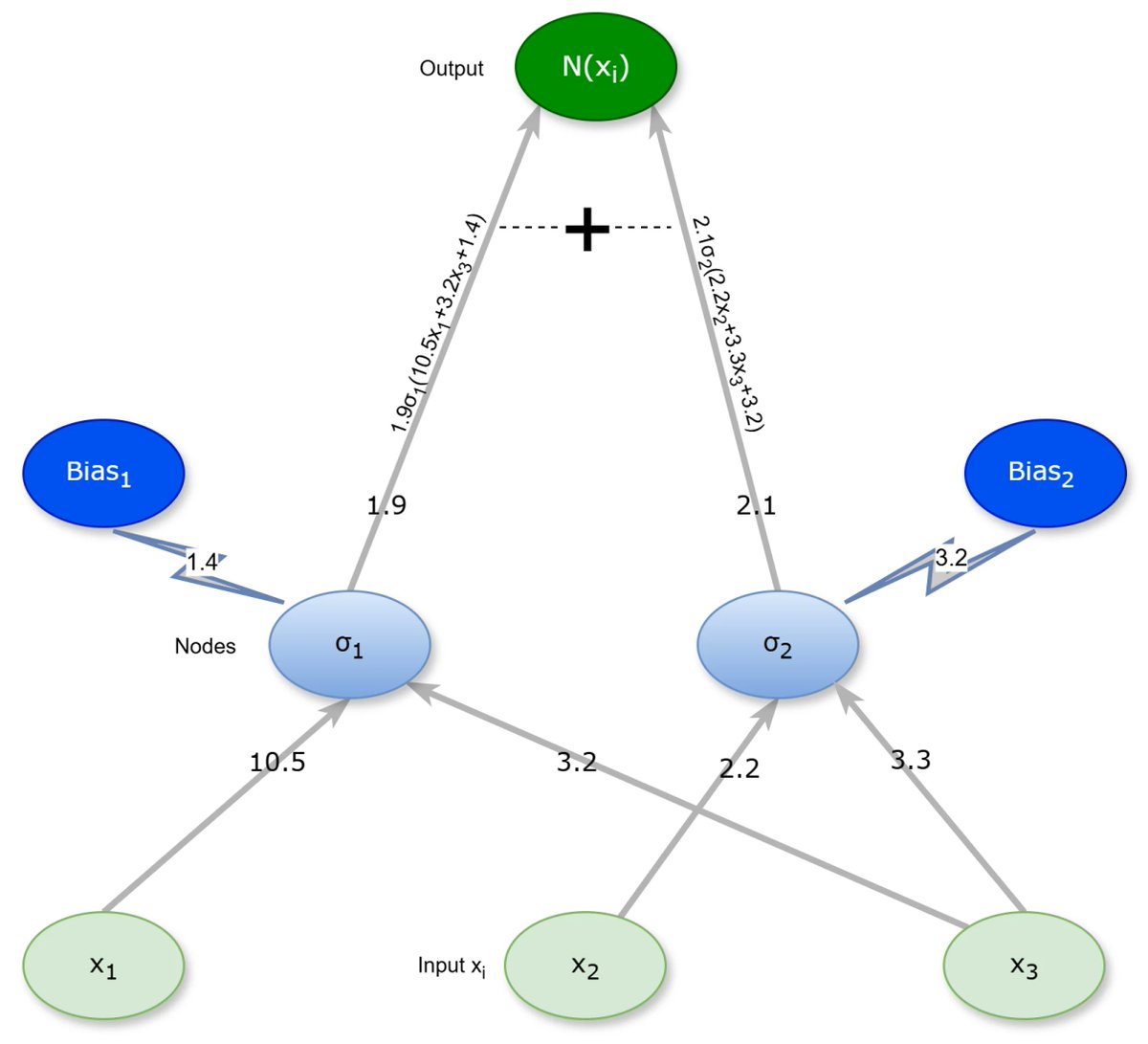
Check this newly published article "Improving the Performance of Constructed Neural Networks with a Pre-Train Phase" at brnw.ch/21x2tpp
Authors: Ioannis G. Tsoulos, Vasileios Charilogis and Dimitrios Tsalikakis
#mdpisymmetry #neuralnetworks #geneticalgorithms
2
2
60
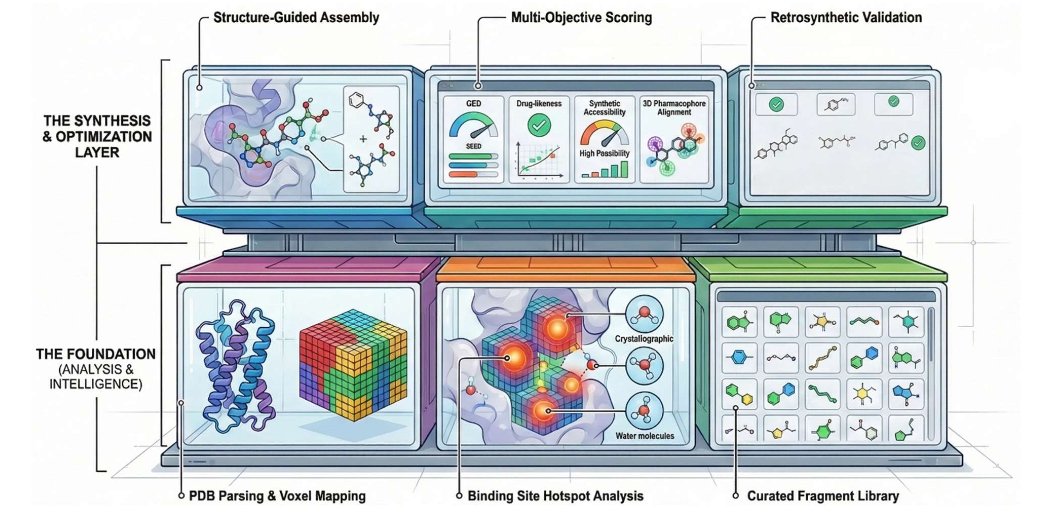
LigandForge: A Web Server for Structure-Guided De Novo Drug Design
1 LigandForge is presented as an end-to-end, browser-based workflow for structure-guided de novo ligand design that explicitly couples pocket physics (3D voxel fields hotspots water thermodynamics) with synthesis awareness (retrosynthetic feasibility), aiming to reduce both licensing and programming barriers.
2 The core technical idea is a voxel-based property grid around the binding site: electrostatics, hydrophobicity, steric accessibility/excluded volume, openness/SASA-derived cavity boundaries, plus geometric descriptors (curvature/shape index). These fields provide directional gradients used to guide fragment placement and growth in 3D.
3 A notable feature is explicit handling of crystallographic waters: each water site is assigned replaceability and entropy/energy-related contributions, and high-energy waters are flagged as displacement targets to guide ligand functionalization with thermodynamic rationale.
4 Molecules are built via chemistry-aware fragment assembly from curated libraries organized by functional role (core scaffolds, linkers, substituents, bioisosteres). An attachment-point manager checks local hybridization (sp/sp2/sp3), aromaticity, and valence, while fragments are oriented using local field gradients to match pocket interaction demands.
5 Drug-likeness constraints are enforced during growth (not only after generation): MW 150–700 Da, LogP −4 to 7, rotatable bond limits, and heavy atom count constraints (10–50). This keeps candidates within practical physicochemical ranges throughout the assembly trajectory.
6 Multi-objective optimization combines pharmacophore/hotspot alignment, QED-based drug-likeness, synthetic accessibility, and novelty/diversity into a weighted composite score with target-class presets (e.g., kinase vs GPCR) that adjust weights for different binding-site archetypes.
7 Optimization supports reinforcement learning, genetic algorithms, and hybrid heuristics; chemical space coverage is maintained using fingerprint-based diversity control with DBSCAN clustering to reduce mode collapse and encourage scaffold variety.
8 Synthesis awareness is integrated as a hard filter/penalty: a retrosynthetic analyzer decomposes top candidates into routes and assigns difficulty, step/yield proxies, and feasibility. The final reward is scaled by route feasibility and penalized by difficulty, pushing optimization away from impractical structures.
9 Prospective evaluation used Boltz-2 co-folding/affinity prediction on three targets (D2R, EGFR, STAT5b-NTD). Across 10 runs per target (100 candidates/target), candidates reached micromolar predicted affinities, with submicromolar predictions for D2R; predicted poses recapitulated key motif interactions (D2R D3.32 salt bridge; EGFR hinge H-bonding in the ATP site).
10 Novelty was assessed against known actives (ChEMBL for D2R/EGFR; limited literature set for STAT5b-NTD): every generated molecule reportedly had Tanimoto similarity < 0.3 to the closest known active for its target, suggesting exploration of distinct chemotypes while retaining predicted binding competence.
💻Code: github.com/HTS-Oracle/Ligand…
📜Paper: biorxiv.org/content/10.64898…
#DeNovoDesign #FragmentBasedDrugDesign #StructureBasedDrugDesign #Cheminformatics #RDKit #ReinforcementLearning #GeneticAlgorithms #Retrosynthesis #WebServer #DrugDiscovery
1
5
39
3,117



1
10
286

6 Nov 2025
A Genetic Algorithm to Scour Protein Sequence Space: A novel framework for protein engineering using protein language models and force fields
1. Researchers introduce GAPO (Genetic Algorithm for Protein Optimization), a groundbreaking framework that integrates genetic algorithms with protein language models and force fields to efficiently explore protein sequence space. This innovative approach outperforms traditional methods like simulated annealing in optimizing protein stability and binding affinity.
2. GAPO leverages evolutionary computing to navigate the vast and complex landscape of protein sequences. By combining genetic algorithms with advanced protein language models such as ESM2, it achieves superior optimization results, demonstrating higher average sequence probabilities and more favorable energy scores compared to wild-type and other optimized variants.
3. The study highlights GAPO's flexibility, allowing users to customize initialization methods, fitness functions, and mutation techniques. This adaptability makes it suitable for a wide range of protein engineering tasks, from optimizing structural stability to enhancing functional properties.
4. In a case study optimizing Hen-Egg Lysozyme, GAPO demonstrated significant improvements in both ESM2 likelihoods and REF15 force field scores. The framework's ability to balance exploration and exploitation of sequence space led to more effective optimization trajectories compared to simulated annealing.
5. GAPO's implementation supports various initialization strategies, including random and seeded initialization with natural or ESM-generated sequences. The results show that ESM-seeded populations explored more diverse regions of sequence space, suggesting a promising approach for discovering novel protein variants.
6. The framework also incorporates advanced selection, crossover, and mutation techniques informed by evolutionary scale modeling. These features enable GAPO to maintain population diversity while efficiently converging towards high-fitness solutions.
7. The study underscores the potential of combining data-driven protein language models with physics-based force fields. This hybrid approach could pave the way for future protein design strategies that integrate the strengths of both methodologies.
8. GAPO's modular architecture allows for easy integration of new models and scoring functions, ensuring its continued relevance as the field of protein engineering evolves. The framework's ability to combine multiple fitness functions opens new avenues for multi-objective optimization.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinEngineering #GeneticAlgorithms #ProteinLanguageModels #ForceFields #Bioinformatics #ComputationalBiology
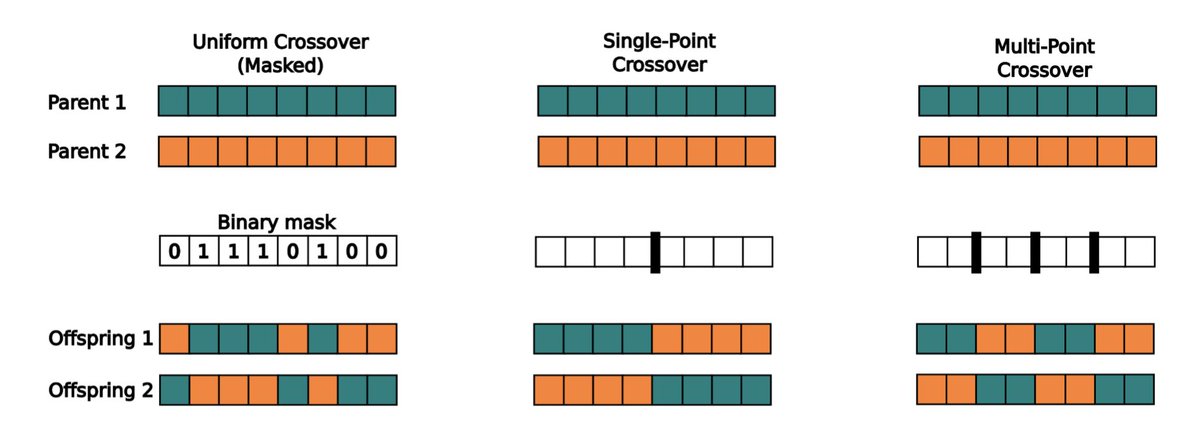
3
867

23 Oct 2025
😲Insufficient charging infrastructure is a huge roadblock for #ElectricVehicles (EV) adoption! Traditional route-planning studies have major flaws, ignoring battery nonlinearity and dynamic factors. But here's the breakthrough🎉An intelligent partial charging strategy using an improved genetic algorithm slashes computational complexity and cuts travel time by 15%!
#geneticalgorithms
Link[doi.org/10.1080/10095020.202…]
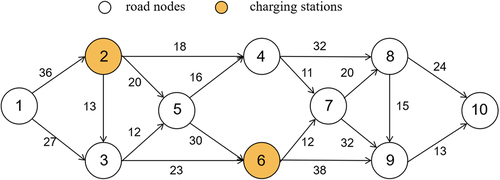
ALT Figure 5. Schematic diagram of a simple road network.
5
227

O‘zbekistonliklarda dunyoda noma’lum bo‘lgan gen o‘zgarishlari aniqlandi
Batafsil👉 fazo.tv/olam-uz/ozbekistonli…
#ozbekiston #uzbekistan #gen #Genetics #geneticalgorithms #olimlar #sciencefiction #sciencenews #interesting #nikoh #marriage #newgen #BREAKING #breakingnews #explore

3
41

2 Oct 2025
Genetic algorithms uncover solutions that brute force would miss, improving everything from shipping logistics to portfolio optimization.
link in comment
#elixir #geneticalgorithms #functionalprogramming

2
2
4
665



18 Aug 2025
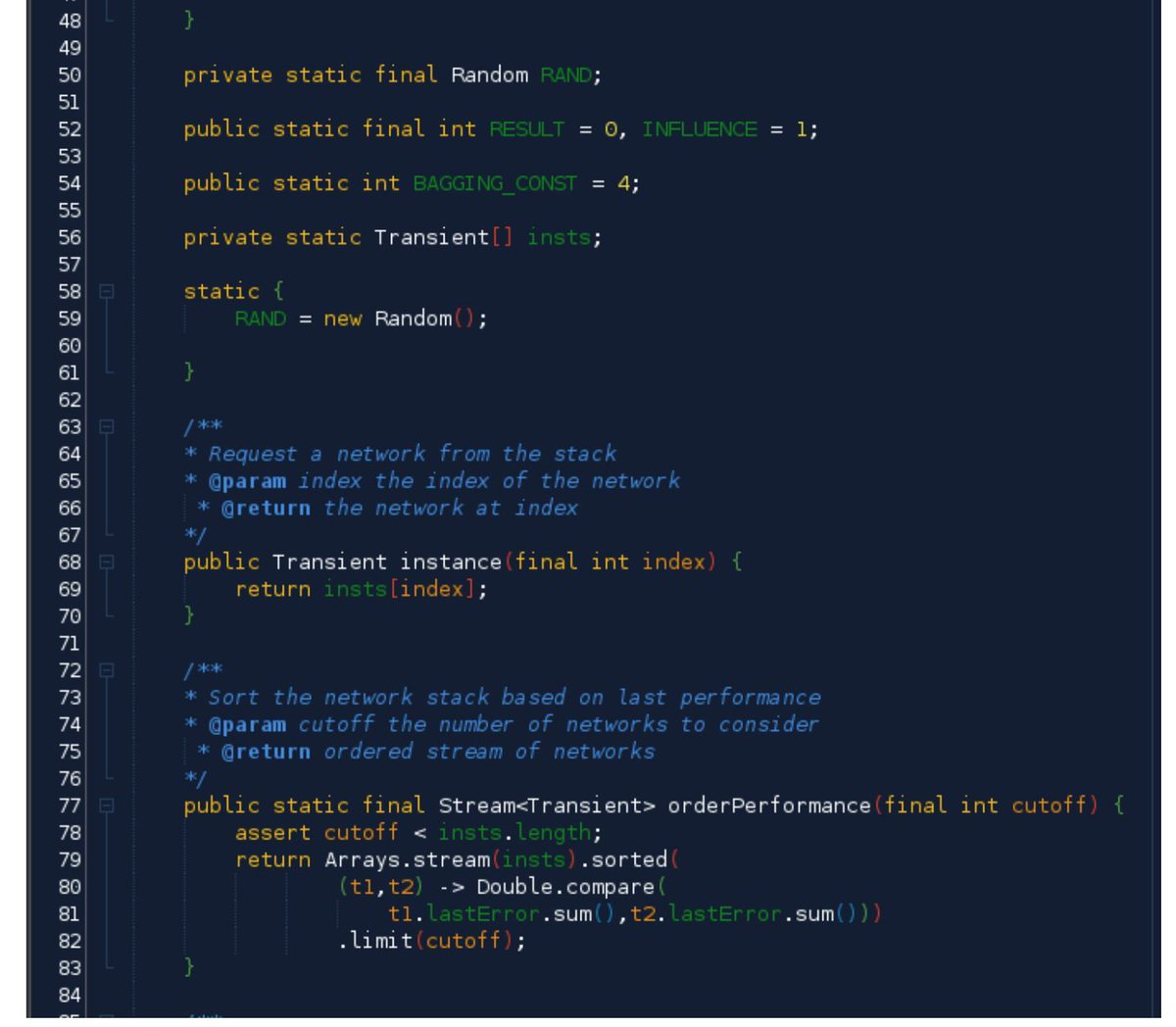
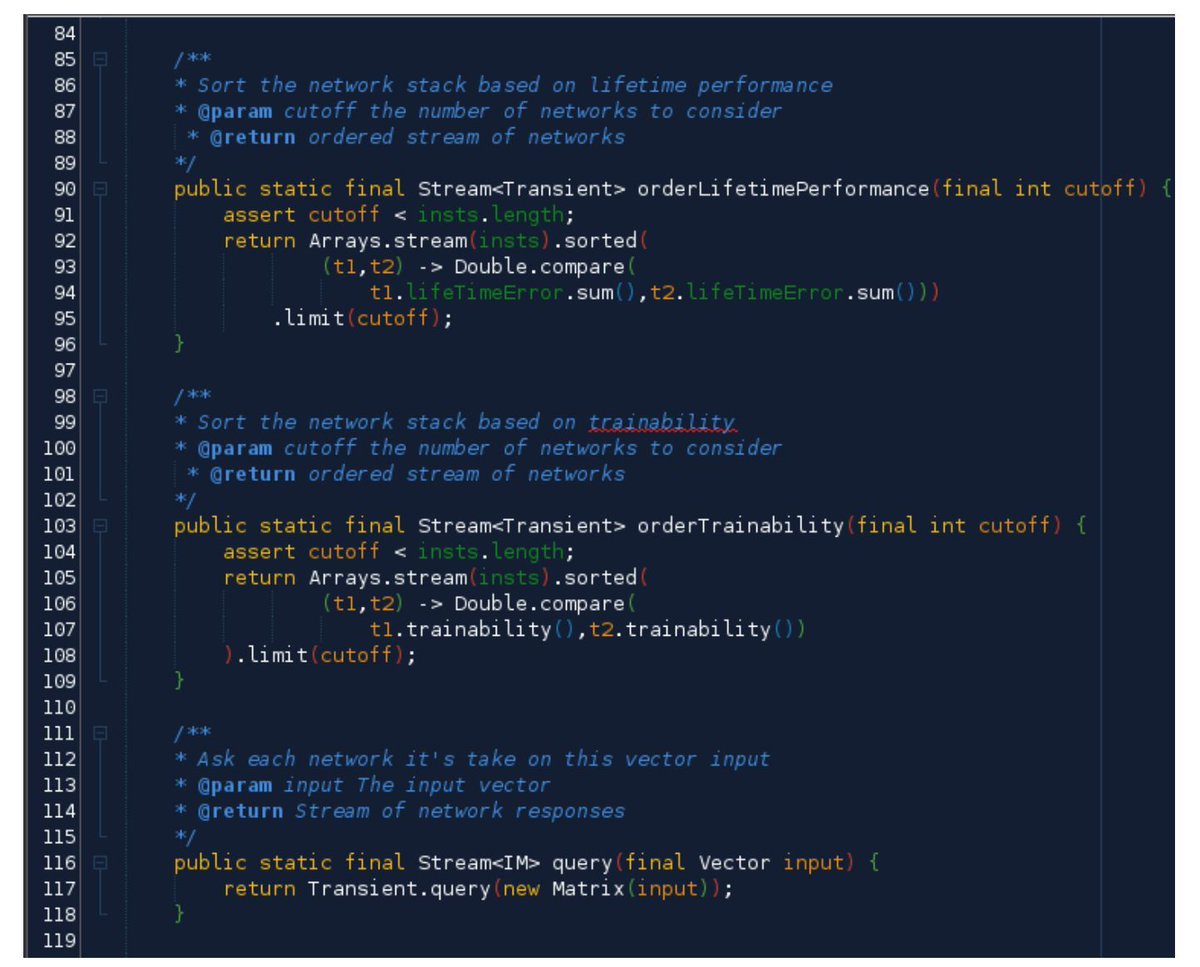
The #GeneticAlgorithms Implementation! #BigData #Analytics #DataScience #AI #MachineLearning #IoT #IIoT #Python #RStats #TensorFlow #JavaScript #Java #GoLang #CloudComputing #Serverless #DataScientist #Linux #Programming #Coding #100DaysofCode
geni.us/JavaImpl
3
2
221

How can #DigitalTwins and #GeneticAlgorithms optimize #BuildingEnergySystems to reduce costs and #CarbonEmissions effectively? Read the full study!
Read more in a research paper published in #SCPE, Vol. 26, No. 3, ISSN 1895-1767: tinyurl.com/4ezmp6h5
2
2
29
26 Jul 2025
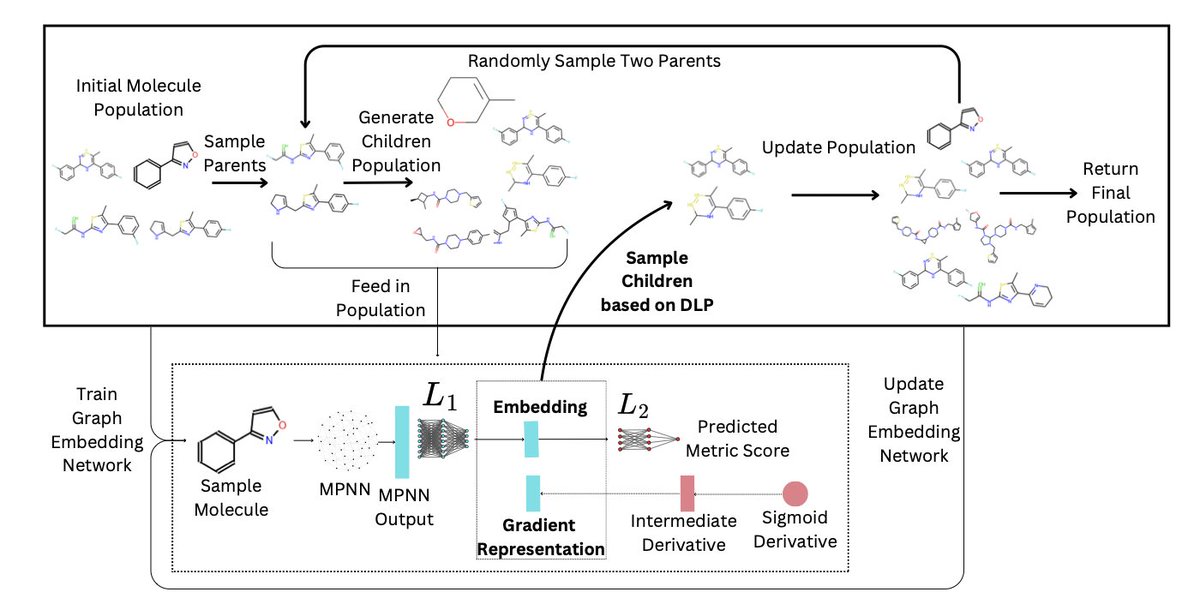
Gradient GA: Gradient Genetic Algorithm for Drug Molecular Design
1. The Gradient Genetic Algorithm (Gradient GA) is a novel approach that integrates gradient information into traditional genetic algorithms to enhance the efficiency and effectiveness of drug molecular design. This method significantly improves convergence speed and solution quality by leveraging gradient guidance in discrete molecular spaces.
2. Gradient GA uses a differentiable objective function parameterized by a neural network and applies the Discrete Langevin Proposal to enable gradient-based exploration. This allows each proposed sample to iteratively progress toward an optimal solution, mitigating the random-walk behavior typical of traditional genetic algorithms.
3. Experimental results demonstrate that Gradient GA outperforms state-of-the-art techniques, achieving up to a 25% improvement in the top-10 score over the vanilla genetic algorithm. The method shows consistent superiority across various molecular optimization benchmarks.
4. The study highlights the potential of combining gradient-based methods with evolutionary algorithms, opening new avenues for more efficient and targeted drug discovery. Future work will explore further integration of gradient information and optimization techniques in this domain.
💻Code: github.com/debadyuti23/Gradi…
📜Paper: openreview.net/pdf?id=G0nfe4…
#GradientGA #DrugDesign #MolecularOptimization #AIinBiology #GeneticAlgorithms
1
2
13
1,379
26 Jul 2025
Gradient GA: Gradient Genetic Algorithm for Drug Molecular Design
1. The Gradient Genetic Algorithm (Gradient GA) is a novel approach that integrates gradient information into traditional genetic algorithms to enhance the efficiency and effectiveness of drug molecular design. This method significantly improves convergence speed and solution quality by leveraging gradient guidance in discrete molecular spaces.
2. Gradient GA uses a differentiable objective function parameterized by a neural network and applies the Discrete Langevin Proposal to enable gradient-based exploration. This allows each proposed sample to iteratively progress toward an optimal solution, mitigating the random-walk behavior typical of traditional genetic algorithms.
3. Experimental results demonstrate that Gradient GA outperforms state-of-the-art techniques, achieving up to a 25% improvement in the top-10 score over the vanilla genetic algorithm. The method shows consistent superiority across various molecular optimization benchmarks.
4. The study highlights the potential of combining gradient-based methods with evolutionary algorithms, opening new avenues for more efficient and targeted drug discovery. Future work will explore further integration of gradient information and optimization techniques in this domain.
💻Code: github.com/debadyuti23/Gradi…
📜Paper: openreview.net/pdf?id=G0nfe4…
#GradientGA #DrugDesign #MolecularOptimization #AIinBiology #GeneticAlgorithms

2
10
1,309



27 Jun 2025
4
6
89
3,335