Khanh M. N. An (光絃之明) retweeted

6 Aug 2025
#DirectedEvolution of Copper-Substituted Nonheme #Enzymes for Enantioselective Alkene Oxytrifluoromethylation by James G. Zhang and Xiongyi Huang (@xiongyih) in @J_A_C_S pubs.acs.org/doi/10.1021/jac…
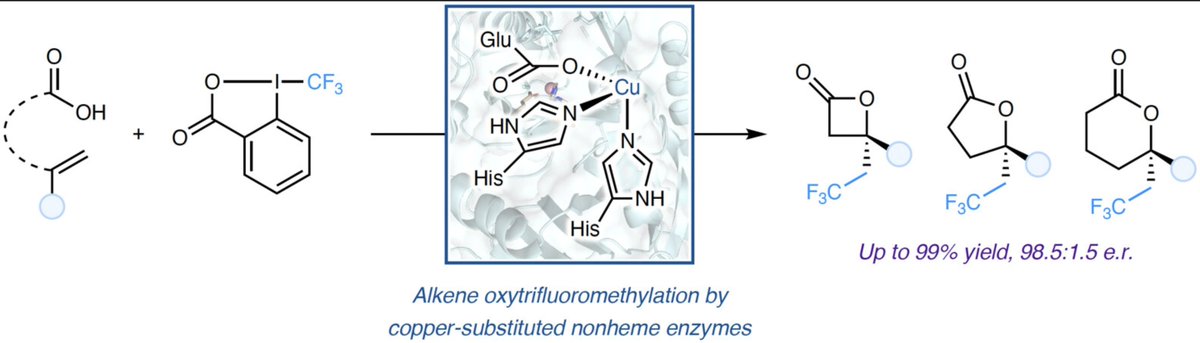
1
12
61
6,074
Heuristic multi-site optimization for protein sequence design using Masked Protein Language Models @PLOSCompBiol
1 ProtHMSO is a heuristic protein sequence design framework that uses masked protein language models (mainly ESM-2) to propose context-aware, multi-site substitutions, aiming to escape local optima and reduce the “invalid/destabilizing” variants common in blind random mutagenesis.
2 Key idea: mask one or multiple target positions, let the ProtLM output substitution probabilities conditioned on the entire sequence context, and use top-k (k=3 worked best) candidate substitutions to generate a small, high-potential mutant set for fitness scoring—shrinking combinatorial search while keeping evolutionary/biophysical plausibility.
3 The multi-site masking is central: substitutions for all masked sites are predicted synchronously from global context, so probabilities update as the sequence changes. This provides a zero-shot way to capture epistasis (synergistic residue interactions) without explicit structural supervision or task-specific training.
4 ProtHMSO is positioned as both (a) a standalone iterative optimizer and (b) a plug-in mutation operator that can replace random exploration steps inside classic search methods, improving convergence and sample-efficiency.
5 GA-HMSO: integrates ProtHMSO into a genetic algorithm by replacing random mutation with ESM-2-guided mutation, and uses a multi-objective fitness (sum of predictor scores). A dynamic schedule (higher mutation early, lower later) improved exploration–exploitation balance and avoided premature convergence.
6 MCTS-HMSO: integrates ProtHMSO into Monte Carlo Tree Search by using ESM-2 probabilities to guide expansion. It also introduces grouping of child nodes by mutation position (choose site first, then substitution), mitigating the wide-and-shallow tree problem in high-dimensional sequence action spaces.
7 AMP benchmark (DBAASP-derived; three challenging cases): across 1–5 site mutations, ProtHMSO consistently improved antimicrobial metrics (PAMP, PMIC) over random mutagenesis, while also improving structural plausibility proxies (higher ESMFold pLDDT, lower ProGen2 perplexity). Notably, random multi-site mutation degraded plausibility as sites increased, while ProtHMSO improved it.
8 ProteinGym benchmark (long proteins; single-site focus for scalability): on the Clinical substitution benchmark, ProtHMSO produced about 2x more non-pathogenic variants than random mutation at matched library sizes (10/50/100 variants per sequence). On DMS benchmarks (including targeted functional-site mutagenesis), ProtHMSO showed higher enrichment of experimentally top-ranked high-fitness mutants (top-10/20/50 overlaps) for both single- and two-site settings.
9 Practical framing: ProtHMSO acts as a high-throughput “candidate narrowing” layer—reducing millions of possibilities to tens/hundreds—while remaining compatible with adding stricter downstream filters (e.g., Rosetta/MD) in a coarse-to-fine pipeline.
💻Code: github.com/chen-bioinfo/Prot…
📜Paper: doi.org/10.1371/journal.pcbi…
#ProteinDesign #ProteinEngineering #ProteinLanguageModels #ESM2 #DirectedEvolution #GeneticAlgorithms #MCTS #AntimicrobialPeptides #ProteinGym #ComputationalBiology

1
25
2,047

Jun 3
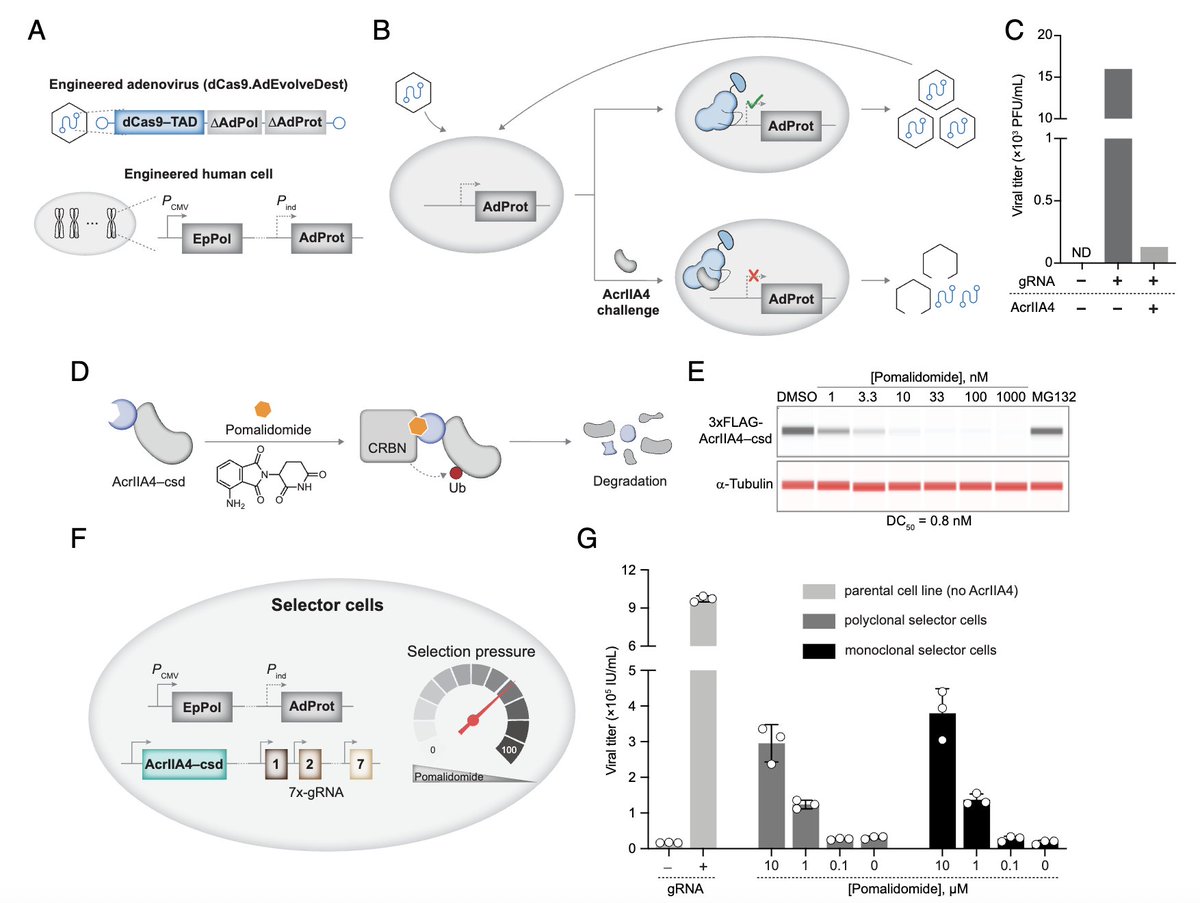
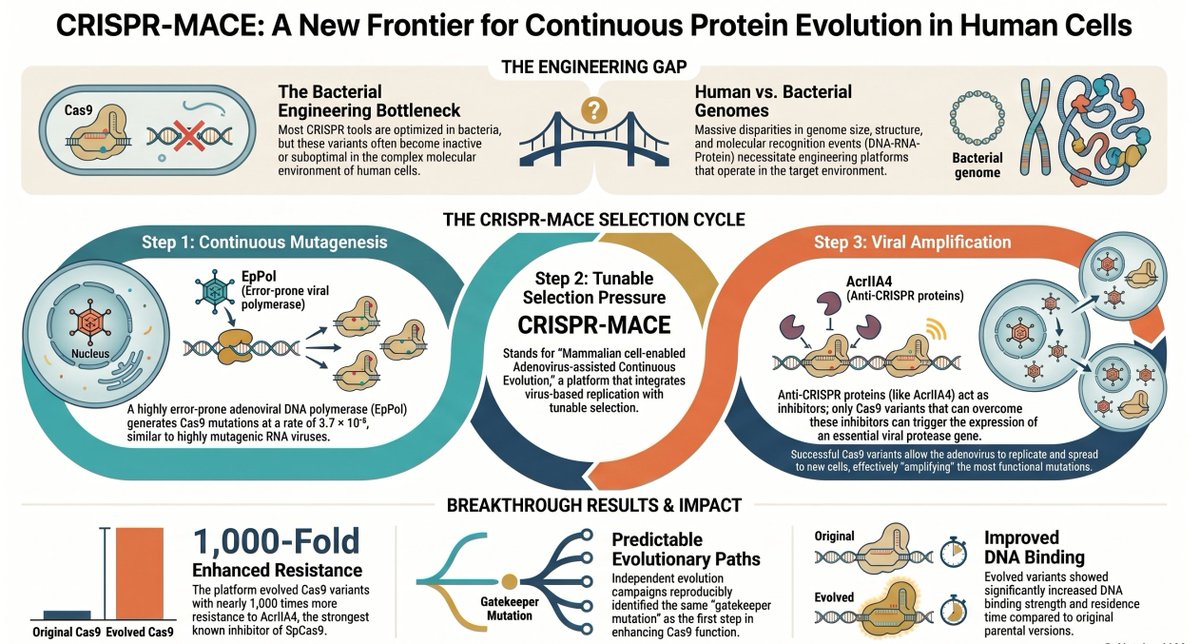
🧬 CRISPR evolution — directly inside human cells — for the first time.
PNAS: CRISPR-MACE uses adenovirus replication to continuously mutate, select, and amplify Cas9 variants in human cells, bypassing bacterial evolution limitations.
Key results:
🔑 "Gatekeeper" G12D mutation: 3× higher DNA affinity
💪 DVKIH 5-mutant variant: ~1000× anti-CRISPR resistance
✅ Validated in CASFISH imaging — brighter, stronger signal
Shoulders & Choudhary labs, Broad Institute | PNAS
doi.org/10.1073/pnas.2536003…
#CRISPR #DirectedEvolution #Cas9 #GeneTherapy #Broadinstitute

1
19
84
6,445
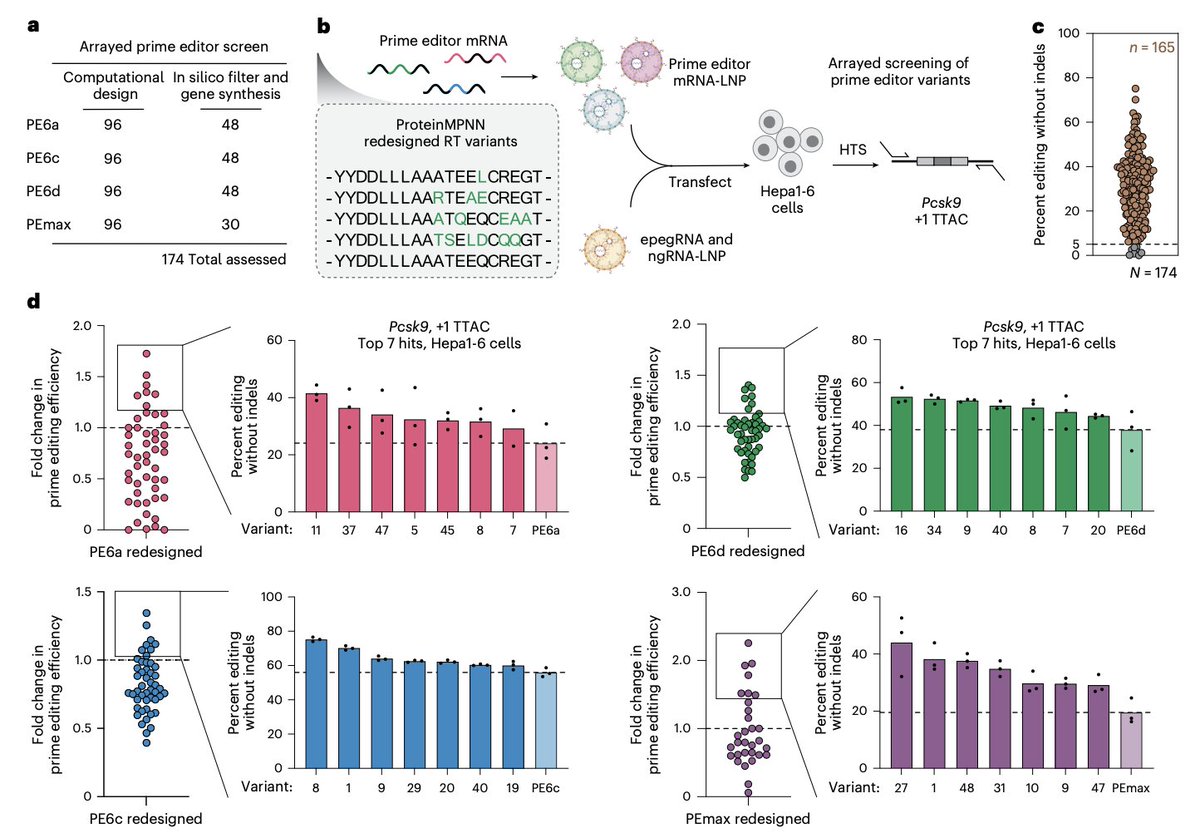
AI-guided redesign of laboratory-evolved reverse transcriptases enhances prime editing @NatureBiotech
1. The study identifies a key bottleneck in state-of-the-art prime editors: reverse transcriptases (RTs) optimized by directed evolution often trade catalytic gains for poorer folding, reduced stability, and lower intracellular expression—limiting performance especially in transient delivery settings.
2. To address this, the authors apply structure-informed AI redesign (ProteinMPNN) to RT domains from multiple prime editor lineages (PEmax, PE6a, PE6c, PE6d), while explicitly preserving residues near substrates and highly conserved positions to protect catalytic function.
3. The redesign strategy is unusually aggressive: redesigned RTs carry 30–163 amino acid substitutions (up to ~24% sequence divergence; up to ~40% of residues allowed to vary in candidate designs), far beyond prior prime editor engineering efforts that typically introduced ≤17 RT substitutions.
4. A practical pipeline is demonstrated: AlphaFold-predicted structures guide ProteinMPNN sequence generation under distance-to-substrate and conservation constraints, followed by AlphaFold2 filtering using pLDDT and RMSD to retain structurally faithful designs before synthesis and screening.
5. In an mRNA-LNP screen (Pcsk9 1 TTAC in Hepa1-6 cells), 95% of redesigned RTs remained functional (>5% editing), and ~30% outperformed their parental editors—showing that broad sequence exploration can still preserve complex RT-dependent prime editing activity.
6. Broad validation uses a pooled “self-targeting” lentiviral assay covering 700 ClinVar pathogenic variants (plus controls), totaling 16,800 prime-edit measurements across redesigned RTs; top redesigned variants improve average editing efficiency across many edits, particularly for heavily evolved RTs.
7. The best redesigned editors are named PE8 variants: PE8a (from PE6a), PE8c (from PE6c), PE8d (from PE6d), and PE8max (from PEmax). Across multiple comparisons, PE8 variants often exceed PE6/PEmax and also outperform PE7 (PEmax–La) in several tested contexts.
8. Mechanistically, redesigned RTs increase soluble expression and stability: intracellular prime editor protein levels after mRNA-LNP delivery rise up to ~2-fold (peak at ~8 h), bacterial soluble RT yields improve, and DSF shows notable thermostability gains for some designs (for example, PE8c RT 8 °C Tm over PE6c).
9. The work emphasizes that higher expression/stability translates into better editing under therapeutically relevant delivery constraints: improvements are shown in primary human fibroblasts, CD34 HSPCs, and primary T cells via mRNA electroporation, and via eVLP RNP delivery.
10. In vivo (mouse liver, Pcsk9 1 TTAC), PE8 variants increase editing up to 2.9-fold versus PE6/PEmax under modest dosing; higher in vivo protein expression accompanies higher editing, while edit:indel ratios and candidate off-target analyses remain comparable to prior editors.
💻Code: github.com/Allentaoyz/Redesi…
📜Paper: doi.org/10.1038/s41587-026-0…
#PrimeEditing #GenomeEditing #CRISPR #ProteinDesign #ProteinMPNN #AlphaFold #DirectedEvolution #mRNADelivery #LNP #ComputationalBiology
1
4
30
2,248
Integrating Diffusion and Liquid AI Models for Predicting Peptide Affinity from mRNA Display Selections
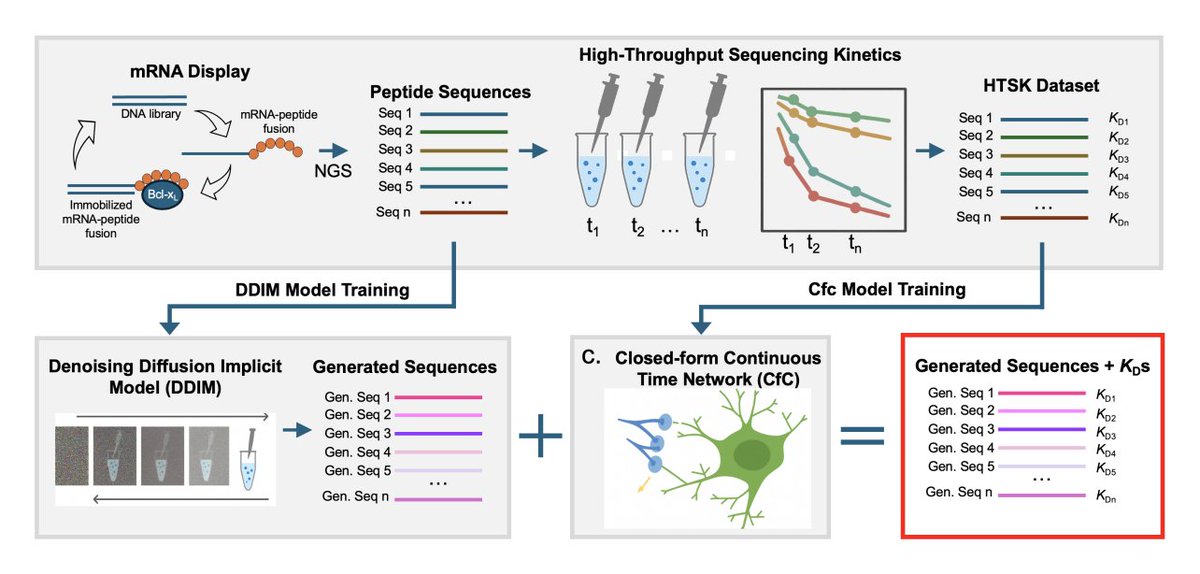
1. The study presents an end-to-end in silico pipeline that couples (i) diffusion-based sequence generation from mRNA display deep sequencing with (ii) a Liquid AI predictor (Closed-form Continuous, CfC) that outputs quantitative binding free energies (ΔG°) for each generated peptide.
2. The biological use case is peptide binding to the oncogenic protein Bcl-xL, leveraging a large, target-specific quantitative dataset from high-throughput sequencing kinetics (HTSK): 15,700 peptides (21-mers) paired with experimentally derived ΔG° values.
3. The key modeling choice is a continuous-time “liquid” neural network (CfC), motivated by the idea that sequence→affinity mapping behaves like an irregular series: amino-acid identity, position, and context produce non-uniform, nonlinear effects, and measured affinities are unevenly distributed across sequence space.
4. CfC is used instead of the earlier Liquid Time-Constant (LTC) networks to avoid expensive numerical solvers: CfC provides a differentiable closed-form dynamics with sigmoid time-gating, reported to yield ~10–100× faster training while retaining accuracy.
5. Model training details: peptides are one-hot encoded as 20×20 matrices; the dataset is split 70:15:15; architecture includes batch norm, dense layers (tanh), two CfC blocks (one returning full sequence, one final state), and a linear output for ΔG°; trained with Adam and MSE for 10 epochs.
6. On held-out HTSK test sequences, predicted ΔG° closely matches the experimental distribution: mean/median both -14.9 kcal/mol; MAE ~0.27 kcal/mol; MSE ~0.12; predictions show mild smoothing and compression mainly at the weaker-affinity end (less represented in training).
7. The diffusion component (DDIM) is trained on selection-derived sequencing data to generate novel functional peptides beyond the experimentally observed pool; 20,000 new sequences are sampled with near-identical positional amino-acid frequency to the training distribution (KL divergence 0.0036).
8. The combined workflow (DDIM generation CfC scoring) is experimentally validated: multiple DDIM-generated candidates predicted to be strong binders are synthesized as mRNA–peptide fusions and kinetically measured; several show single-digit picomolar affinities, including ~4-fold improvements over the E1 reference peptide.
9. Quantitatively, across 14 kinetically evaluated DDIM-generated peptides (DDIM-HTSK1–4 and FG variants), CfC predictions agree well with experiments (reported MSE < 0.4 kcal/mol); importantly, the model can rank close sequence neighbors differently when affinity differs substantially (e.g., FG7 vs DDIM-HTSK4).
10. Limitations are clearly demonstrated: when DDIM proposes mutations in an extremely conserved motif region (e.g., positions 10–15 “Y-KAAD” where 99.9% of training sequences match), CfC may still predict high affinity, yet pull-down experiments show little binding—highlighting out-of-distribution and low-sampling failures, and the need for broader dynamic range and coverage.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #ProteinEngineering #PeptideDesign #mRNADisplay #DirectedEvolution #MachineLearning #DiffusionModels #LiquidNeuralNetworks #BindingAffinity #BclxL
7
16
1,552
Simple Baselines Rival Protein Language Models in Mutation-Dense Design Tasks
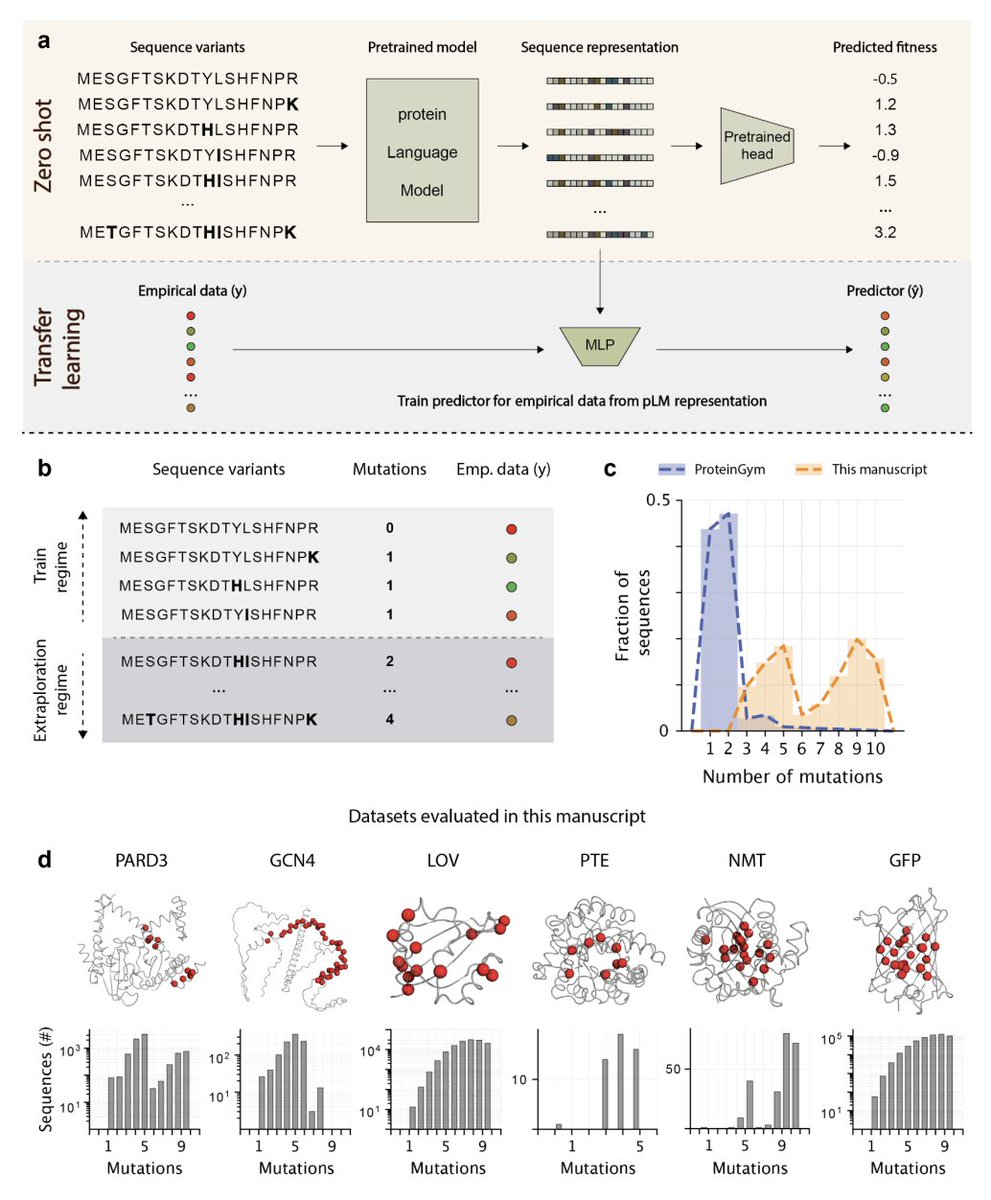
1. The study benchmarks popular protein language models (pLMs) specifically in mutation-dense, high-order combinatorial design landscapes, where the goal is to extrapolate from low-order measured mutants to unseen higher-order combinations (a design-relevant OOD setting often missing from standard benchmarks).
2. Across six dense multi-mutant datasets (FuncLib/htFuncLib: GFP, PTE, NMT; ProteinGym: LOV, PARD3, GCN4), zero-shot pLM scoring showed weak separation of functional vs non-functional variants (often near-chance ROC AUC ~0.5–0.6) and modest phenotype correlations (Spearman ρ ranging roughly from negative to moderate positive values).
3. A key baseline result: zero-shot pLM scores were statistically indistinguishable from a simple homology-based evolutionary prior (PSSM). In other words, for ranking/triage in these combinatorial active-site regimes, pLMs did not consistently outperform “classical” sequence statistics computed from an MSA.
4. For transfer learning, the authors introduce an extrapolation-controlled protocol: train on variants up to mutational order k_train and test on a fixed higher order k_test, explicitly quantifying performance degradation with increasing train–test mutational gap (rather than using only random splits).
5. In transfer learning across eight pLMs (ESM2 sizes from 8M to 3B, ProtBert, ProGen2 variants), model identity/scale/architecture contributed little: pLMs were largely indistinguishable from each other, and in ~96% of pLM-to-pLM comparisons differences were not statistically significant.
6. The dominant driver of predictive performance was the train–test mutation gap (how far the test variants are from what was measured), not parameter count or training cost. Notably, ESM2-8M could match or exceed ESM2-3B in these extrapolative regimes despite ~1000x fewer parameters.
7. A second central baseline: in supervised prediction, a simple one-hot encoding (OHE) of substitutions paired with the same downstream MLP rivaled—and often matched or outperformed—the best pLM representation, both under controlled extrapolation and under standard random train/test splits across training set sizes.
8. Beyond prediction, the paper tests model-guided landscape exploration: ranking beneficial vs deleterious single mutations and nominating combinatorial “neighborhoods” to define screening libraries enriched for gain-of-function variants. pLMs again did not significantly outperform PSSMs on mutation ranking or library enrichment metrics.
9. The authors’ interpretation: pretraining on natural sequences may encode “tolerated” evolutionary variation but is not a reliable standalone signal for gain-of-function combinatorial design, especially under strong epistasis typical of active sites. They argue pLMs may need explicit biophysical/structural priors or integration with structure-based approaches to improve design utility.
10. Practical contribution: the mutation-dense datasets used here are deposited to ProteinGym to support future benchmarking/training, and the paper emphasizes that any new “general-purpose” design claim should be tested against strong, simple baselines (PSSM, OHE) under extrapolation-controlled splits.
💻Code: github.com/drprfitay/fitness…
📜Paper: biorxiv.org/content/10.64898…
#ProteinDesign #ProteinLanguageModels #MachineLearning #ComputationalBiology #Benchmarks #Epistasis #DirectedEvolution #Bioinformatics #Rosetta #ProteinEngineering
3
14
46
3,938
Towards a Generative Protein Evolution Machine with DPLM-Evo
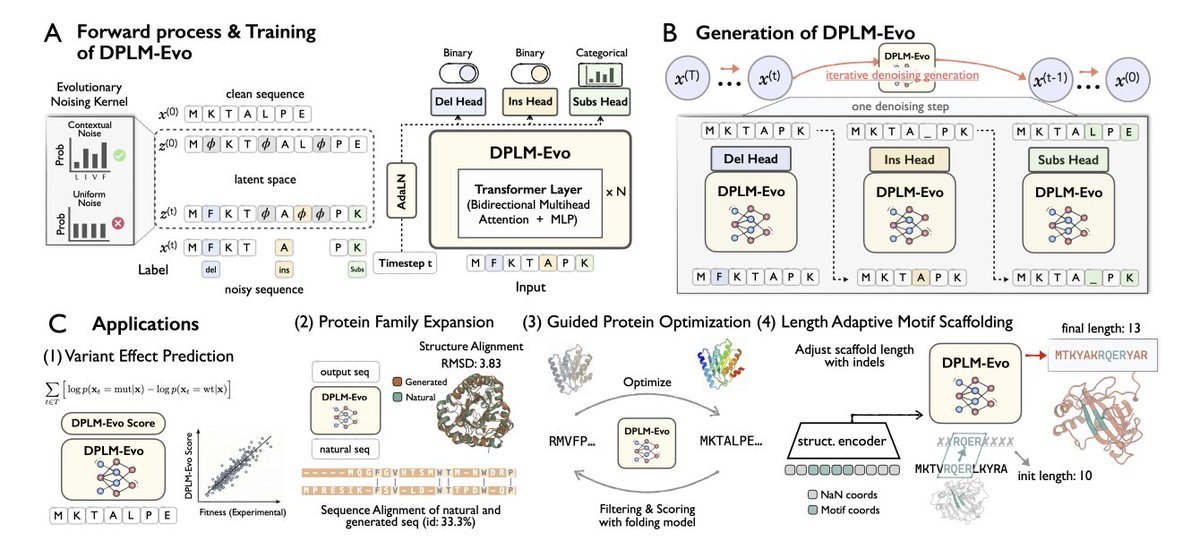
1. DPLM-Evo reframes protein diffusion generation around explicit evolutionary edits: substitutions, insertions, and deletions (indels). This addresses a mismatch in prior DPLMs where proteins “emerge from masks,” despite real evolution proceeding via accumulated edits.
2. Core idea: decouple a fixed-size latent alignment space from the variable-length observed sequence. Indels become gap ↔ residue transitions in the latent space, making variable-length diffusion tractable while keeping compute close to fixed-length models.
3. Architecture: the model denoises in observed sequence space but predicts three edit signals per position via separate heads: (i) amino-acid distribution for substitution, (ii) deletion probability, and (iii) insertion probability (insert to the right; residue identity comes from the substitution head).
4. A key innovation is the contextualized evolutionary noising kernel for substitutions. Instead of uniform random corruption, substitutions are corrupted using a context-dependent distribution derived from the model’s own predictions (after a warmup), producing more biologically plausible mutation patterns during training.
5. This contextualized corruption materially matters: an ablation replacing it with uniform corruption drops ProteinGym average Spearman from 0.42 to 0.295; a static BLOSUM-based kernel lands in-between (~0.35), supporting the claim that context-aware mutation noise better matches evolutionary constraints.
6. Understanding task highlight: DPLM-Evo achieves state-of-the-art mutation effect prediction on ProteinGym among single-sequence foundation models (217 DMS assays). Scoring is “substitution-native”: it directly reads substitution probabilities at mutated sites without masking them, avoiding an artificial mask-token scoring mismatch.
7. Indel effect prediction: on the ProteinGym indel benchmark, DPLM-Evo reaches 0.495 Spearman, outperforming strong single-sequence baselines (e.g., ProGen2 M 0.464) and approaching MSA-based methods (PoET 0.517, ProFam ensemble 0.530), suggesting explicit indel modeling transfers to indel fitness estimation.
8. Generation: DPLM-Evo enables variable-length unconditional protein generation via evolutionary denoising (sub/ins/del), starting from a learned prior rather than an all-mask state. It maintains strong foldability (ESMFold pLDDT ~83.6, comparable to DPLM) while improving diversity and reducing repetition/mode collapse.
9. Conditional design: in motif scaffolding, DPLM-Evo can dynamically adjust scaffold length during sampling (via insertion/deletion heads) while keeping motif residues fixed, avoiding manual enumeration of scaffold lengths required by fixed-length generators; it improves solved motif counts and success rate in zero-shot and further with continued finetuning.
10. Edit-trajectory applications: the model supports post-editing and optimization as explicit evolutionary trajectories. Case studies include in-silico “family expansion” (large sequence divergence while preserving fold) and directed evolution of GFP, where enabling indels improved structural scores faster and higher than substitution-only and an ESM-2 baseline under the same search/filtering protocol.
📜Paper: arxiv.org/abs/2605.00182
#ComputationalBiology #ProteinDesign #ProteinLanguageModels #DiffusionModels #GenerativeAI #MachineLearning #Bioinformatics #DirectedEvolution #ProteinEngineering #VariantEffectPrediction
1
6
26
2,415
Fitness Translocation: Improving Variant Effect Prediction with Biologically-Grounded Data Augmentation
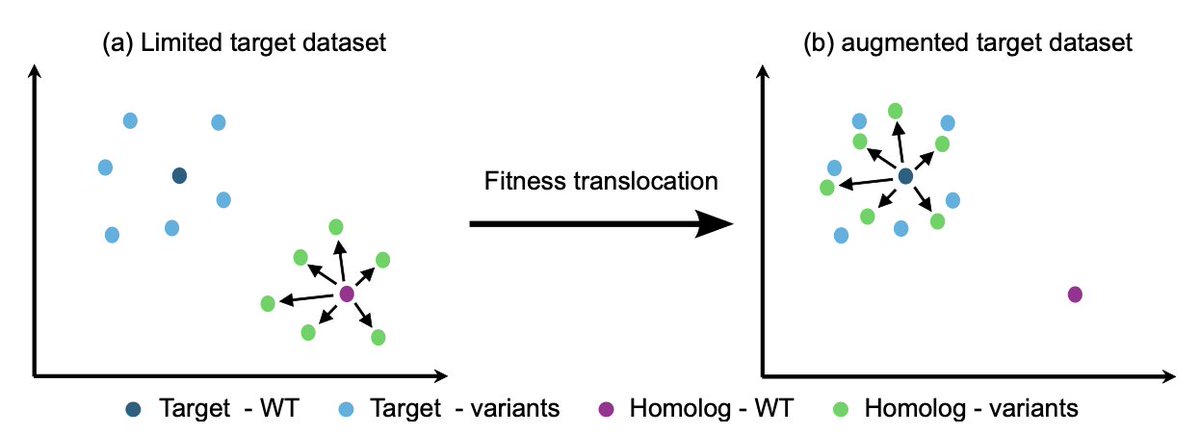
1. The study tackles the long‑standing bottleneck of sparse protein fitness data, which hampers accurate mapping from sequence to function in engineering and evolution.
2. It introduces *fitness translocation*, a data‑augmentation strategy that transfers the mutational effect observed in a homologous protein to a target protein, thereby enriching the target’s training set without new experiments.
3. Using pretrained protein language models, the method computes an embedding offset for each homolog variant (variant embedding minus wild‑type embedding) that captures the mutation’s directional change in latent space.
4. The offset is applied to the target wild‑type embedding, creating a synthetic variant that inherits the homolog’s measured fitness (normalized by wild‑type fitness), thus preserving biological relevance in the augmented data.
5. Because the technique operates solely in embedding space, it requires no sequence alignment and can be applied to homologs with as little as 35 % sequence identity, making it well suited for low‑data regimes.
6. The authors benchmarked fitness translocation on three diverse protein families—IGPS enzymes, green fluorescent proteins, and SARS‑CoV‑2 spike proteins—using multiple predictors (SVR, RF, Lasso) and language models (ESM‑2, ESM‑1v).
7. Across all configurations, augmentation consistently improved Spearman correlation, with the largest gains observed for SARS‑CoV‑2 spike cell‑entry predictions, followed by IGPS enzymatic activity and GFP fluorescence.
8. A homolog‑selection algorithm, grounded in one‑sided paired t‑tests, identifies which homologs yield statistically significant performance boosts, preventing the inclusion of noisy or irrelevant data.
9. Ablation studies show that removing either the statistical test or the sequential selection stage degrades results, underscoring the algorithm’s role in achieving robust improvements.
10. Principal‑component analysis demonstrates that translocation aggregates homolog variant embeddings around the target, indicating that mutational impacts are effectively transferred across sequence space.
11. The method’s success aligns with evidence that fitness landscapes are conserved across phylogenetically distant proteins, validating the biological assumption that evolutionary pressures preserve functional constraints.
12. By expanding usable training data, fitness translocation can accelerate directed evolution and generative protein design, potentially reducing the number of costly experimental cycles needed to reach high‑performance variants.
💻Code: github.com/adrienmialland/Pr…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinEngineering #MachineLearning #ProteinLanguageModels #VariantEffectPrediction #DataAugmentation #DirectedEvolution #ComputationalBiology #SARSCoV2 #IGPS #GFP #Bioinformatics
3
13
1,574
FLIP2: Expanding Protein Fitness Landscape Benchmarks for Real-World Machine Learning Applications
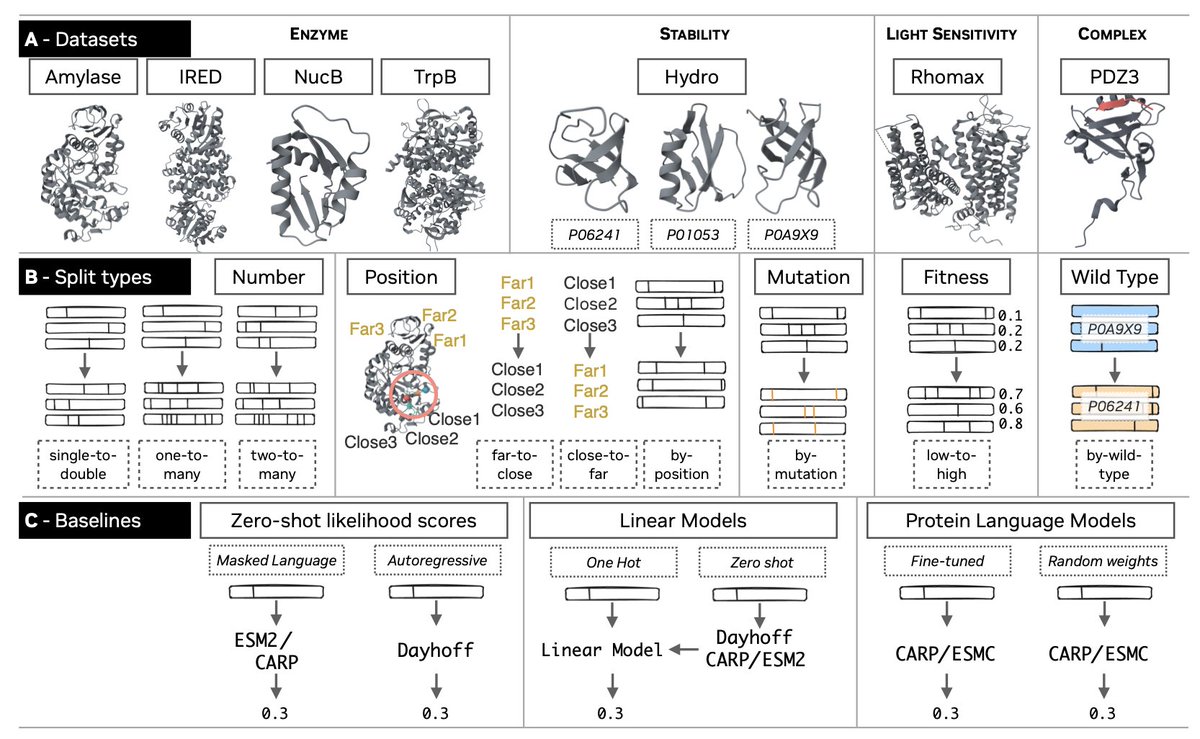
1. The FLIP2 benchmark introduces seven new sequence-fitness datasets spanning enzymes, protein-protein interactions, and light-sensitive proteins—significantly broadening functional diversity beyond previous benchmarks that were limited to thermostability, binding, and viral capsid viability.
2. A key innovation lies in the novel split strategies designed to mimic real-world protein engineering constraints: generalization across wild-type backgrounds, extrapolation to unseen mutations and positions, and prediction of higher-fitness variants from lower-fitness training data.
3. Surprisingly, extensive evaluations reveal that simpler models often matched or outperformed fine-tuned protein language models on FLIP2, challenging the prevailing assumption that transfer learning techniques always provide superior performance for fitness prediction.
4. The benchmark systematically tests zero-shot likelihood scores from protein language models (Dayhoff, CARP, ESM2), ridge regression on one-hot encodings, and fine-tuned pLMs—revealing that no single approach dominates across all generalization scenarios.
5. Critical findings show that current pLM architectures struggle with wild-type and position splits, indicating fundamental limitations in generalizing across protein scaffolds and to previously unobserved mutational positions—precisely the settings most relevant to practical engineering campaigns.
6. The datasets include industrially relevant proteins: alpha-amylase for detergent applications, imine reductase for pharmaceutical production, NucB nuclease for chronic wound care, and rhodopsin variants for optogenetics—ensuring benchmark relevance to biotechnology applications.
7. All data are redistributed under CC-BY 4.0 license with full provenance documentation, establishing a rigorous, reproducible foundation for advancing machine learning methods in protein engineering.
💻Code: flip.protein.properties
📜Paper: biorxiv.org/content/10.64898…
#proteinengineering #machinelearning #benchmark #fitnesslandscape #proteinlanguagemodels #syntheticbiology #directedevolution #computationalbiology
9
46
2,855
PRIZM: Combining Low-N Data and Zero-shot Models to Design Enhanced Protein Variants
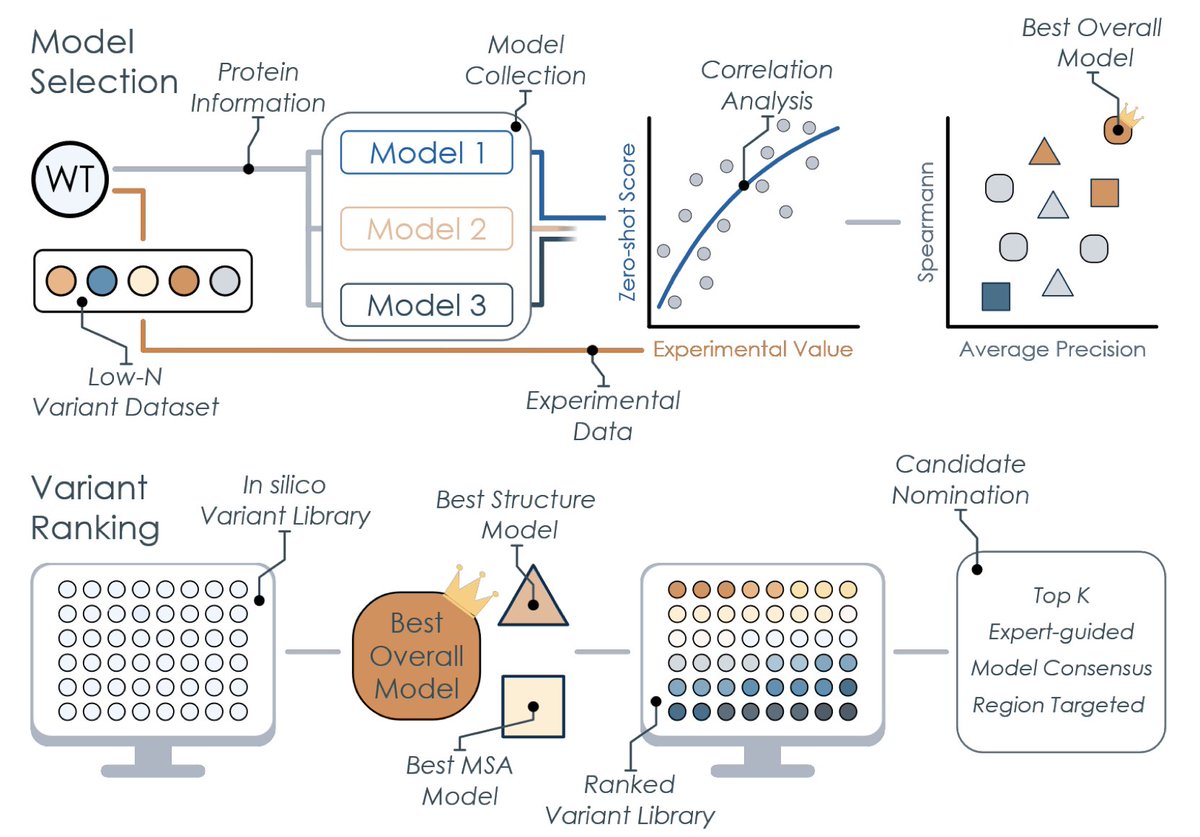
1 PRIZM introduces a practical two-phase workflow that bridges the gap between expensive supervised learning and blind zero-shot prediction in protein engineering.
2 The method requires only ~20 labeled variants to identify the best-performing zero-shot foundation model for a specific protein property, making it accessible to labs without ML expertise.
3 Unlike traditional ML-assisted directed evolution, PRIZM leverages existing experimental datasets rather than demanding new unbiased training data, reducing experimental burden.
4 In a sucrose synthase thermostability case study, PRIZM identified a variant with ~3°C higher melting temperature and 60% residual activity at 60°C versus 23% for wild-type.
5 For a glycosyltransferase activity optimization with just 8 initial variants, PRIZM achieved a 60% hit rate for improved variants, discovering beneficial mutations distant from the active site that rational design would likely miss.
6 The approach evaluates 25 diverse zero-shot models spanning sequence, structure, and MSA-based architectures, selecting those most correlated with experimental data rather than relying on global benchmark averages.
7 PRIZM addresses a critical limitation in current zero-shot workflows: model performance varies dramatically across different proteins and properties, making task-specific model selection essential.
8 The workflow outputs ranked variant libraries without requiring model training, finetuning, or specialized computational infrastructure, democratizing access to foundation model-guided protein design.
💻Code: github.com/daha-la/PRIZM
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #MachineLearning #DirectedEvolution #ZeroShotLearning #Bioinformatics #EnzymeEngineering #SyntheticBiology #ComputationalBiology
6
22
1,538
Efficient, Few-shot Directed Evolution with Energy Rank Alignment
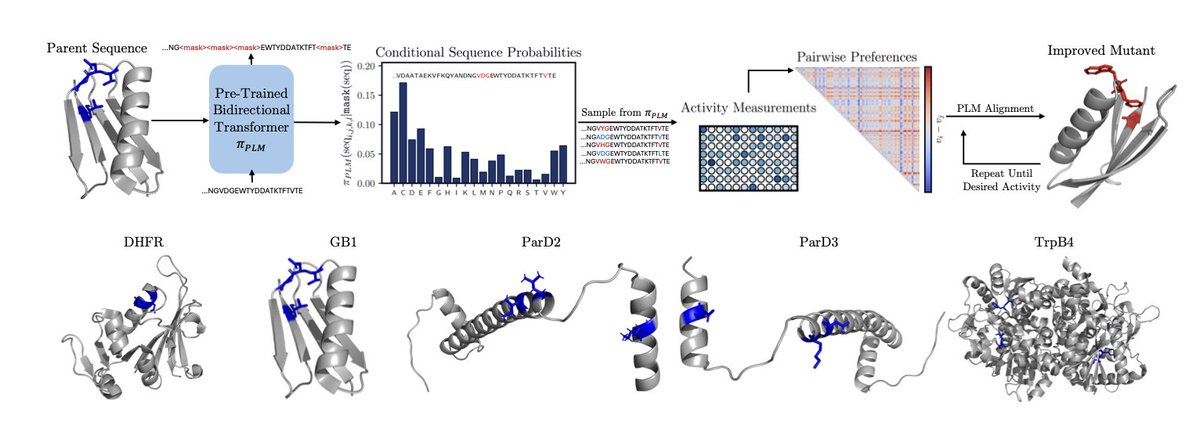
1. A new method called Energy Rank Alignment (ERA) enables highly efficient protein engineering by adapting large pre-trained protein language models using minimal experimental data.
2. Unlike previous approaches that rely on simple models due to sparse data constraints, ERA leverages the strong inductive biases of ESM3-1.4B, a 1.4 billion parameter protein language model, to navigate complex fitness landscapes.
3. The key innovation is using quantitative experimental rankings rather than just binary preferences, allowing the model to preserve relative fitness magnitudes while learning from small batches of just 96 sequences per round.
4. ERA outperforms existing methods including MLDE, ALDE, EVOLVEpro, and Direct Preference Optimization across five diverse combinatorial fitness landscapes involving antibiotic resistance, antibody binding, and enzymatic activity.
5. The method achieves state-of-the-art performance with only 384 total samples across four rounds, successfully finding global optima even in landscapes with strong epistatic effects and rugged topography.
6. Surprisingly, adding structural conditioning or thermostability pre-training did not improve performance, suggesting that pure sequence-based adaptation is sufficient for effective directed evolution.
7. The adapted models maintain sequence diversity while shifting probability mass toward high-fitness regions by several orders of magnitude, making them interpretable and useful for understanding biophysical requirements.
8. This work establishes a compelling interface between foundation models and experimental design, demonstrating how post-training algorithms from statistical physics can solve real biological optimization problems.
💻Code: github.com/rotskoff-group/er…
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #DirectedEvolution #MachineLearning #Bioinformatics #ProteinDesign #ESM3 #ComputationalBiology #FewShotLearning
2
11
52
2,871
Repurposing Protein Language Models for Latent Flow-Based Fitness Optimization
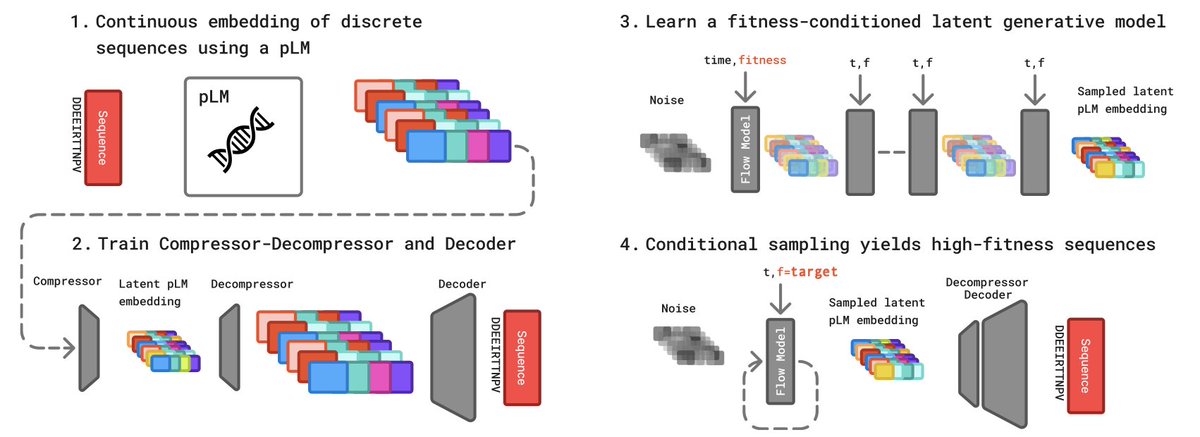
1 CHASE turns a frozen protein language model into a high-fitness sequence generator by compressing ESM2 embeddings into a 32-length latent code and training a flow-matching model conditioned on fitness alone—no predictor gradients at sampling time.
2 The key trick is classifier-free guidance in the compressed latent space: drop fitness labels 20 % of the time during training, then extrapolate with a guidance scale w to steer sampling toward any target fitness in a single ODE solve of 40 steps.
3 Benchmarked on the standard AAV & GFP “Medium/Hard” landscapes, CHASE reaches 0.62/0.61 (AAV) and 0.91/0.92 (GFP) median normalized fitness, outperforming VLGPO, GGS, GPE* and other strong baselines while keeping diversity and novelty on par.
4 Inference is 10–85× faster than predictor-guided VLGPO because the flow net needs only one forward pass per step; memory stays under 2.4 GB for 128 sequences.
5 A self-bootstrapping loop augments the sparse training set with 25 % synthetic sequences sampled from the same model; retraining lifts AAV Medium fitness from 0.62 → 0.65 and AAV Hard 0.61 → 0.63 without extra wet-lab data.
6 Ablation shows aggressive compression (20×) beats milder 16×, and two-stage VAE training (decoder first, compressor second) stabilizes the latent manifold, giving consistently higher final fitness across all four tasks.
7 The entire pipeline is predictor-free during generation: only a lightweight oracle is used post-hoc to rank top-128 candidates, making CHASE readily deployable on low-data protein engineering campaigns beyond AAV/GFP.
💻Code: github.com/eth-siplab/CHASE
📜Paper: arxiv.org/abs/2602.02425
#proteindesign #flowmatching #proteinLM #directedevolution #computationalbiology #bioinformatics #AIforScience
4
37
2,285
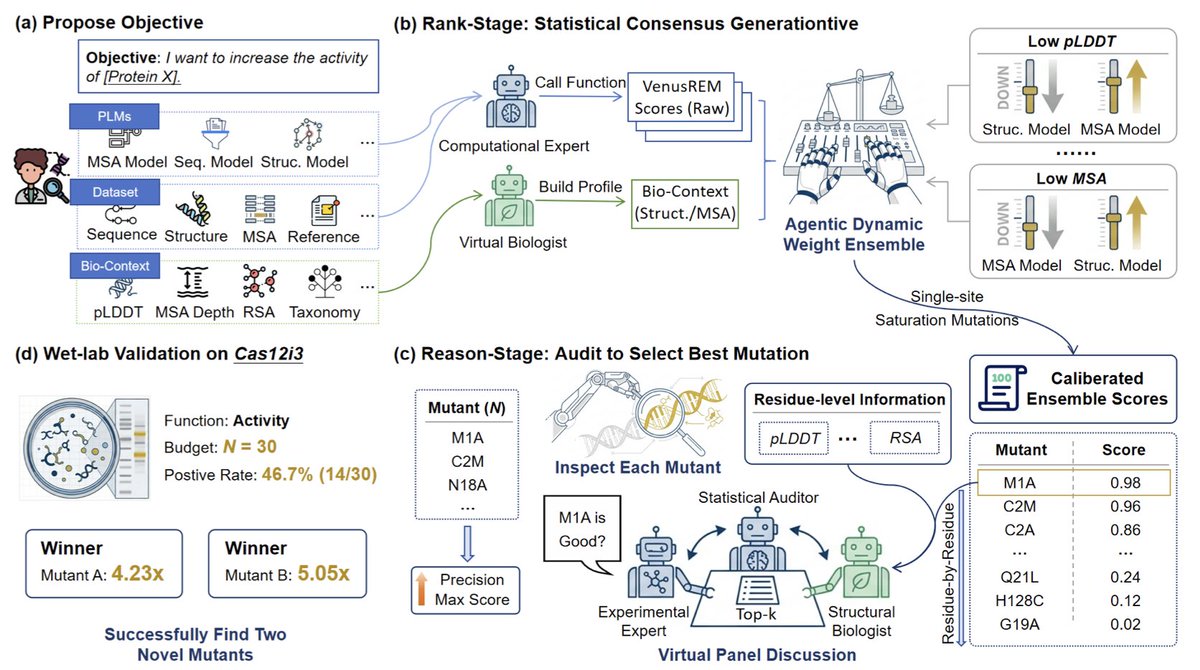
Rank-and-Reason: Multi-Agent Collaboration Accelerates Zero-Shot Protein Mutation Prediction
1 New SOTA on PROTEINGYM: a 0.551 Spearman ρ beats the prior best 0.518 by combining protein language models with a LLM-driven multi-agent panel instead of hand-tuned ensembles.
2 Two-stage agentic pipeline: Rank-stage fuses sequence/structure/MSA scores via a Computational Expert Virtual Biologist that auto-calibrates weights; Reason-stage deploys a Virtual Expert Panel (statistical, structural, wet-lab auditors) to veto biophysically implausible hits.
3 Top-5 hit rate jumps 367 % on the high-coverage PROTEINGYM-DMS99 subset under tight N=30 budgets, showing the gap between global correlation and real selection precision is closed by inference-time chain-of-thought auditing.
4 Wet-lab proof on Cas12i3 nuclease: 14/30 variants improve activity (46.7 % success), two mutants reach 5.05× and 4.23× WT cleavage, validating the framework’s zero-shot utility for CRISPR enzyme engineering.
5 Framework is backend-agnostic: users can hot-swap any PLM or LLM; open prompts and modular code let labs plug proprietary predictors into the same auditing protocol.
💻Code: github.com/ai4protein/VenusR…
📜Paper: arxiv.org/abs/2602.00197
#ProteinEngineering #ZeroShotLearning #MultiAgentSystems #CRISPR #DirectedEvolution #Bioinformatics
5
27
1,914
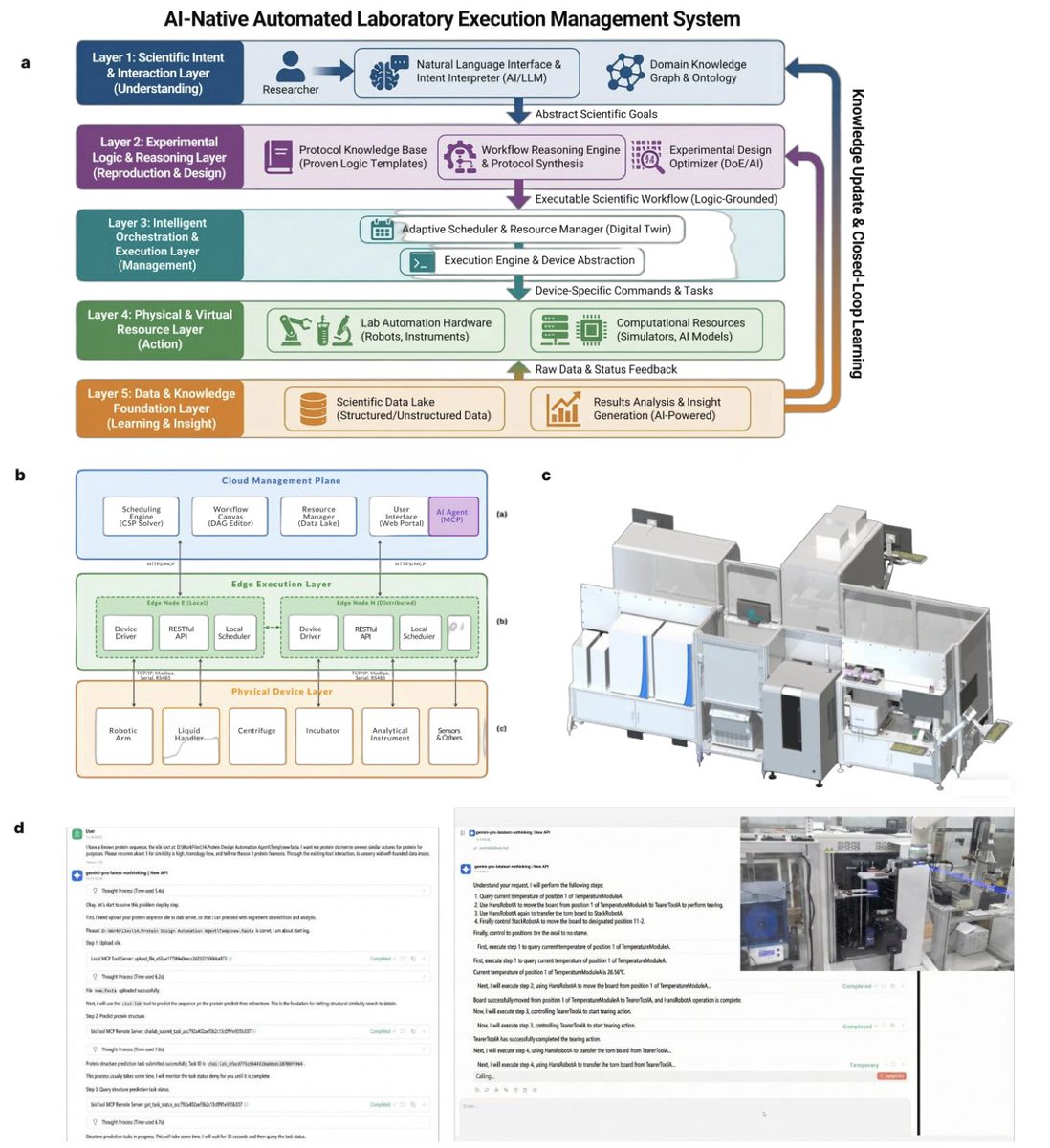
An AI-Native Biofoundry for Autonomous Enzyme Engineering: Integrating Active Learning with Automated Experimentation
1 A “cloud-edge synergistic” biofoundry is presented in which an LLM agent (Qwen3) directly perceives instrument states and actuates wet-lab workflows without human scripts, turning spoken goals into executable protocols.
2 The platform closes the Design-Build-Test-Learn loop in three autonomous rounds, evolving a Family B DNA polymerase for CoolMPS chemistry while sustaining >66 % hit rate and defying the usual law of diminishing returns.
3 Zero-shot ESM-2 priors jump-start exploration from a low-activity scaffold; an EVOLVEpro active-learning model then retrains on each round’s assay data, lifting prediction Pearson r from 0.27 to 0.75 and enriching epistatic anchors such as E485L.
4 Physics-informed mining of 160 thermophilic archaeal homologs identified A0A2Z2MPY8, a natural generalist that outperforms the wild-type in G/C incorporation and serves as an optimal parental backbone for downstream mutagenesis.
5 Automated multi-site mutagenesis, expression, lysis and fluorescence-coupled 3’-N3-dNTP incorporation assays are orchestrated on an MGISP-Smart8 liquid handler, enabling 96-well throughput with minimal manual touch points.
6 Final variant DP1 (E485L R709P) achieves 37 % lower error rate (0.17 % vs 0.27 %) and higher Effective Spot Rate (73 % vs 66 %) than commercial polymerase on DNBSEQ-E25, validating translational utility for antibody-based CoolMPS sequencing.
7 Hardware abstraction via Meta-Action protocol renders devices brand-agnostic; new instruments are hot-plugged without downtime, offering a scalable, vendor-neutral blueprint for future self-driving biotech labs.
💻Code: github.com/MGI-tech/AI-Biofo… (to be released)
📜Paper: biorxiv.org/content/10.64898…
#AIbiofoundry #enzymeengineering #directedevolution #proteinLM #activelearning #CoolMPS #selfdrivinglab
1
3
24
1,522

Our CEO, Michael @Heltzen, was interviewed by @maxinehoovernyc from The Beryl Consulting Group at the @BerylElites’ Investment Conference in New York City:
"Artificial evolution is our way of describing how we can brute force evolution. With the development of AI algorithms over the last couple of years, we have gained access to the capability of foreseeing what a change in the DNA code means to the structure and function of enzymes. So we can basically start designing how the chemical reactions are happening. That means we're expanding the chemical space of what's possible to build, like natural products and drug candidates that were considered impossible to build before."
Interview available here:
na2.hubs.ly/H02Xmfn0
#exozymes #exozyme #enzymes #enzyme #biomanufacturing #cellfree #AI #ML #artificialintelligence #artificialevolution #directedevolution #evolution

2
8
266

28 Dec 2025
Project Veritas
·
26. tammikuuta 2023
**BREAKING:**
tutkii ”mutatoituvan” COVID-19-viruksen käyttöä uusien rokotteiden kehittämiseen
”Älä kerro tästä kenellekään… Tässä on riski… tämän täytyy olla hyvin hallittua, jotta varmistetaan, ettei tämä mutatoitu virus luo jotain… kuten virus sai alkunsa Wuhanissa, rehellisesti sanottuna.”
#DirectedEvolution
Robert F. Kennedy Jr
Uskomaton raportointi James O’Keefelta, joka näyttää meille sen, minkä jo tiesimme: Pfizer on halpamainen, ahne, tappava, moraalisesti vararikossa oleva rikollinen yritys, joka on kaapannut ja turmellut valvojansa.
6
34
1,389

16 Dec 2025
Next-gen CRISPR enables precise gene edits; DE-GE combo boosts crop traits by crafting functional mutations. #CRISPR #DirectedEvolution @OxfordJournals
Details: doi.org/10.1093/hr/uhaf203

5
14
910

22 Nov 2025
Boosting In-Silicon Directed Evolution with Fine-Tuned Protein Language Model and Tree Search
1. A novel framework named AlphaDE is proposed to optimize protein sequences by integrating fine-tuned protein language models and Monte Carlo tree search (MCTS). This innovative approach significantly outperforms previous methods in directed evolution tasks, demonstrating superior efficiency and effectiveness.
2. AlphaDE fine-tunes protein language models using masked language modeling on homologous protein sequences, activating the evolutionary plausibility for the target protein class. This step provides strong prior guidance for the subsequent tree search, enhancing the model's ability to explore the protein fitness landscape.
3. The MCTS inference step in AlphaDE effectively evolves proteins with evolutionary guidance from the fine-tuned model. It balances exploitation and exploration to avoid local optima, making it highly suitable for complex protein fitness landscapes.
4. Extensive benchmark experiments show that AlphaDE remarkably outperforms state-of-the-art methods, even with few-shot fine-tuning. This highlights its potential for practical applications where only limited homologous sequences are available.
5. AlphaDE also demonstrates the ability to computationally condense the protein sequence space, evolving truncated sequences into functional proteins with fewer residues than the wild-type. This opens new possibilities for understanding protein evolution and designing novel proteins.
6. The study highlights the compatibility of AlphaDE with different protein language models, such as ProtBert and ESM-1b, showing its versatility in various protein families. This flexibility makes it a powerful tool for diverse protein engineering tasks.
📜Paper: arxiv.org/abs/2511.09900
#ProteinEngineering #DirectedEvolution #AIinBiology #ProteinLanguageModels #MonteCarloTreeSearch

4
25
2,034
30 Oct 2025
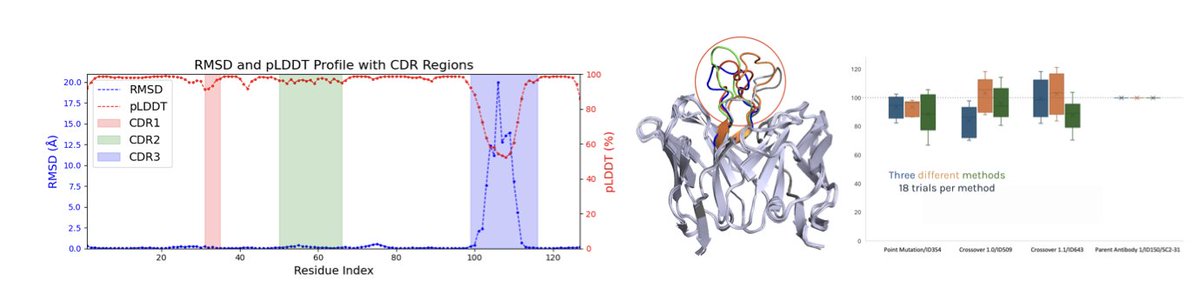
Conformational Rank Conditioned Committees for Machine Learning-Assisted Directed Evolution
1. This paper introduces a novel framework called Rank-Conditioned Committee (RCC) for Machine Learning-assisted Directed Evolution (MLDE) in antibody optimization. The RCC framework leverages ranked conformations to assign a deep neural network committee per rank, enabling a principled separation between epistemic uncertainty and conformational uncertainty. This is a significant advancement in addressing the challenge of modeling conformational uncertainty in antibody design.
2. The authors validate the RCC approach on SARS-CoV-2 antibody docking, demonstrating substantial improvements over baseline strategies. The results show that RCC can more effectively prioritize binding candidates in silico compared to traditional single-structure docking, offering a scalable route for therapeutic antibody discovery, especially against rapidly evolving pathogens.
3. The study integrates ensemble docking with ImmuneBuilder and applies machine learning models within a Bayesian active learning framework. This integration allows for more accurate predictions of antibody-antigen interactions by considering both the uncertainty from model predictions and the structural variability of antibodies.
4. The RCC framework is designed to balance exploration and exploitation in the search for high-affinity antibody variants. By using rank-conditioned acquisition functions, the method can reliably identify promising candidates while avoiding over-penalization of candidates whose uncertainty arises primarily from structural variability.
5. The experimental results highlight the effectiveness of the RCC approach. The mean binding score of the population consistently increases across multiple runs, with the DNN ensemble showing the highest mean shift and reasonable variance, indicating both high exploration and exploitation efficiency.
6. The authors acknowledge that further refinement of the final scoring with more detailed binding simulations and additional wet-lab experimental validation is needed to fully characterize the real-world utility of the approach. However, the current findings suggest that RCC is a promising method for accelerating the discovery of therapeutic antibodies.
📜Paper: arxiv.org/abs/2510.24974
#MachineLearning #DirectedEvolution #AntibodyDesign #ProteinEngineering #ComputationalBiology
3
22
2,313

The iDEC Festival is happening this weekend at the University of Cambridge!✨
Over 200 participants from around the world will present their exciting projects — can’t wait to see what surprises they bring! 🔬🌍
#iDEC2025 #iDEC5Years #SyntheticBiology #DirectedEvolution

2
4
615