Huge thanks to all my brilliant co-authors and mentors 🙌🚀
If you're at ICLR and want to chat AI Agents, Multimodal Learning, Graph Learning, CUAs, or anything else — please stop by our posters or DM me!
See you in Rio 🇧🇷🌊⛱️
#AI #Agents #MultimodalAI #GraphLearning #LLM #CUA
2
1
5
117
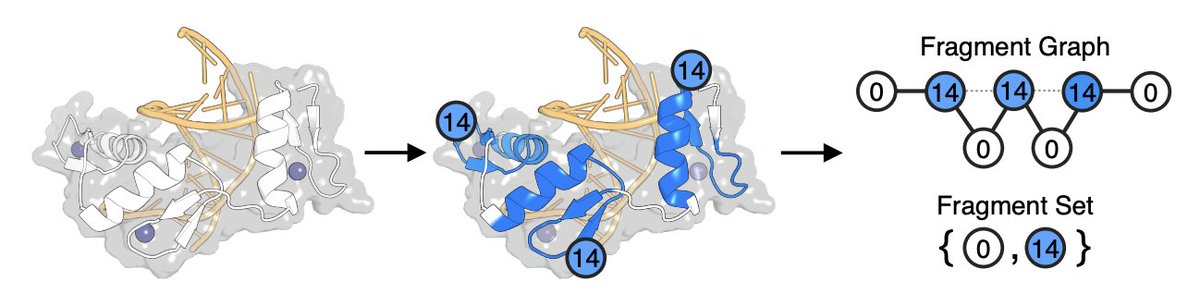
From Atoms to Fragments: A Coarse Representation for Efficient and Functional Protein Design
1. The paper proposes a sparse, interpretable protein representation built from a curated alphabet of 40 evolutionarily conserved “ancient” structural fragments, aiming to replace scaling-heavy sequence or full-atom structure encodings for search and design.
2. Two complementary encodings are introduced: Fragment Sets (presence/absence of fragment types, ignoring arrangement) for speed-critical tasks, and Fragment Graphs (fragments as nodes; peptide-bond and spatial-proximity edges) to retain structural context needed for clustering and design.
3. Fragment detection is performed directly from backbone geometry using a sliding-window scan against a fragment library, evaluating several distance metrics; combining two torsion-angle metrics (LogPr RamRMSD) yields strong detection performance (F1 ≈ 0.85), with an empirically selected classification threshold (3.65%) and AUROC ≈ 87%.
4. On the fold-balanced PDBench benchmark, fragments cover ~40% of residues on average and exhibit distinct biophysical patterns: more intra-fragment hydrogen bonding (notably in mainly-β folds, ~ 15%), fewer inter-fragment hydrogen bonds (notably in mainly-α folds, ~-47%), and slightly reduced solvent accessibility (~-5%), consistent with fragments behaving as more “self-contained” structural units.
5. To test functional signal retention, the authors curate a Protein Function Dataset (PFD) of 215 monomeric proteins spanning 12 binding-function categories (DNA/RNA/ATP/GTP/metal and combinations) filtered to ≤30% sequence identity, making functional grouping challenging for standard similarity measures.
6. Fragment-based distances produce more information-dense embeddings than sequence (BLOSUM) or global shape alignment (RMSD): after PCoA, BagOfNodes (Fragment Sets) preserves >95% variance within 20 dimensions and GraphEditDistance (Fragment Graphs) >80%, vs <60% (BLOSUM) and <40% (RMSD).
7. Functional clustering improves with fragments in multiple ways: BagOfNodes yields very strong cluster compactness/separation (Silhouette ≈ 0.82), while GraphEditDistance best aligns clusters with functional labels (ARI ≈ 0.046; F1 ≈ 0.20), suggesting a practical tradeoff between ultra-compact “bag” features and context-aware graph structure.
8. For functional database search, fragment representations dramatically reduce “tokens per protein” (memory/data points): ~99% fewer than atom/backbone representations and ~94–98% fewer than residue-level sequence representations, while achieving retrieval quality comparable to RMSD/BLOSUM across functions (AUROC/NDCG broadly similar, with some function-specific wins per method).
9. Speed benchmarks (100 queries vs a 100-protein database, 35 cores) show the practical payoff: Fragment Sets (BagOfNodes) answer in ~0.07 s, compared with ~36.6 s for BLOSUM and ~1717 s for RMSD; Fragment Graph edit distance is slower than sequence but still far faster than RMSD (~573 s vs ~1717 s), with a one-time preprocessing cost to build fragment representations.
10. Fragments are also used as functional “blueprints” for generative design: detected fragment backbones are held fixed as templates and RFDiffusion fills missing regions; functional recovery is assessed by FoldSeek hits and GO-code agreement, with reported recovery rates often >40% and reaching near-perfect recovery for some classes (e.g., metal-binding), while random “naive fragments” largely fail—supporting that evolutionary fragment choices, not arbitrary geometry, drive functional signal.
💻Code: github.com/wells-wood-resear…
📜Paper: doi.org/10.1093/bioinformati…
#ProteinDesign #ComputationalBiology #Bioinformatics #ProteinStructure #MachineLearning #DiffusionModels #ProteinSearch #GraphLearning #StructuralBiology #RepresentationLearning
2
7
79
4,675

Mar 25
📢 Call for Papers — COMPLEX NETWORKS 2026
📍 Granada, Spain | Dec 2–4, 2026
Join a leading conference in #NetworkScience & #ComplexSystems
🗓️ Deadline: Sept 2, 2026
🔗 cmt3.research.microsoft.com/…
#CallForPapers #GraphLearning
1
12
276

Feb 27
🎓 PhD Position in Graph Learning
📍 University of Vienna
We seek a motivated PhD student with strong interest in machine learning, graph theory & mathematical foundations.
🗓 Deadline: 26.03.2026
👉 Apply here: jobs.univie.ac.at/job/Univer…
#PhD #GraphLearning #Hiring
2
17
35
1,995

Jan 18
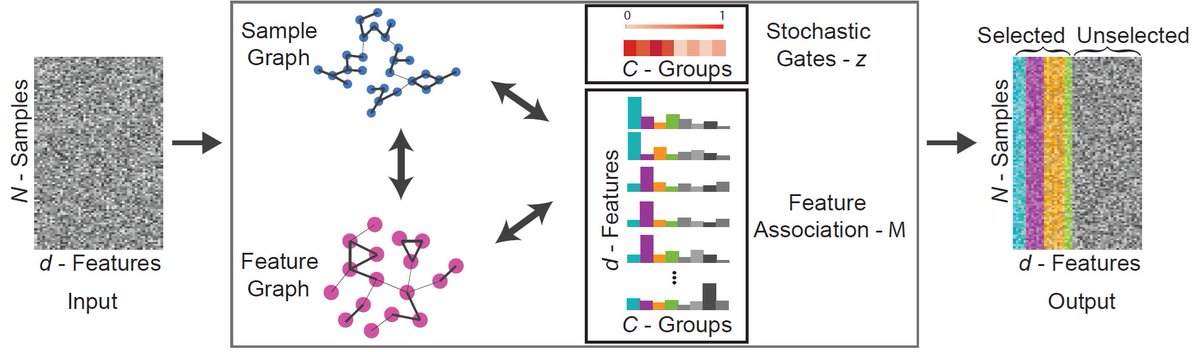
1/
I am thrilled to share that Shira Lifshitz will present our paper at AAAI (oral poster)!
This was a collaborative effort with Ron Meir (EE, Technion), @gmishne (UC San Diego), and @Ofirlin (BIU).
#AAAI #UnsupervisedLearning #FeatureSelection #GraphLearning #DataScience
1
2
4
469

26 Nov 2025
Excited to be heading to NeurIPS 2025 next week in San Diego to present the work I did during my internship at Amazon 😄
If you’ll be around, let’s catch up and chat!
Paper link: openreview.net/forum?id=gBGa…
#NeurIPS #graphlearning
1
5
312

28 Oct 2025
🌧️ PhD alert: Dynamic Graph Learning for Flood-Resilient Cities 🌍
How do urban systems interact during floods?
Apply now: iapetus.ac.uk/studentships/m…
Based in @EngineeringNCL | @Sage_NCL
#PhD #AI #FloodResilience #GraphLearning #UrbanSystems #Hydrology

2
2
98

🚀 Call for Papers – 5th Workshop on Graphs and more Complex structures for Learning and Reasoning (GCLR) is being held in conjunction with AAAI 2026. (aaai.org/conference/aaai/aaa…)
We are fast approaching the deadline for the 5th Workshop on GCLR.
🧩Workshop Format: Fosters collaboration among researchers exploring complex graph structures and reasoning methods — from theoretical models to real-world AI applications.
🧠 Topics of Interest include:
🔹Fairness-aware Learning in Complex Graphs
🔹Benchmarking Foundation Models with Complex Data
🔹Privacy Preservation in Complex Graphs
🔹Causal Inference and Complex Networks
🔹Knowledge Graph-enhanced Foundation Models
🔹Theoretical analysis of graph algorithms or models
🔹Network representation learning and manifold embedding methods
🔹Optimization methods for graphs/manifolds
🔹Link analysis/prediction, node classification, clustering for complex graph structures
🔹Probabilistic and graphical models for structured data
🔹Knowledge graph construction
🔹Social network analysis and measures
🔹Constraint satisfaction and programming (CP), (inductive) logic programming (LP and ILP)
Website: sites.google.com/view/gclr20…
Submission portal: cmt3.research.microsoft.com/…
Submission Guidelines: aaai.org/conference/aaai/aaa…
📅 Important Dates:
Oct 22, 2025 (Pacific Time): Poster/Short/Position Paper Submission Deadline
Oct 22, 2025 (Pacific Time): Full Paper Submission Deadline
Nov 5, 2025: Paper Notification
#AAAI2026 #GCLR2026 #AIResearch #GraphLearning #KnowledgeGraphs #TrustworthyAI #MachineLearning #WSAI #IITMadras
@ravi_iitm @RealAAAI @OfficialINDIAai @rbc_dsai_iitm @IBSE_IITM @ai4bharat @wcte_iitm @cerai_iitm @iitmadras @EduMinOfIndia

4
6
472

4 Oct 2025
Join us for the Stanford Graph Learning Workshop 2025!
🗓️Oct 14, 2025
📍Stanford University
🧠Topics: Agents, RFMs & LLM Inference.
Save your spot to explore the future of #AI, #LLMs and #GraphLearning with leading experts.
Register now: snap.stanford.edu/graphlearn…

3
43
293
17,619

23 Sep 2025
🚀 Excited to share our paper got accepted at DiffCoAlg@NeurIPS 2025 @diffcoalg !🎉
🙏 Thanks to @shrutimoy, Binita Maity, Anant Kumar, @adagu & @cse_iitgn .
#NeurIPS2025 #GNN #GraphLearning #AIResearch

2
6
507

20 Aug 2025
At @cp_conf our coordinator Sylvie Thiébaux delivered an invited talk on Graph Learning for Planning, highlighting how graph-based methods can advance heuristic search in automated planning. #Planning #AI #Graphlearning #TUPLESAI
👉bit.ly/419Xahk

4
71

5 Aug 2025
🚨 BREAKTHROUGH in Graph Learning!
What if each node in your graph could plan, reason, and act like a mini-agent—powered by an LLM? 🤯
That’s exactly what ReaGAN does. And it might just outsmart classic GNNs.
Let me explain 🧵👇
#AI #GraphLearning #LLM #MachineLearning

1
3
74

5 Aug 2025
What if each data point in a network had its own brain? 🤔
A new paper on "ReaGAN" is reimagining graph learning. Traditional methods (GNNs) are powerful but can struggle to see the bigger picture in complex data.
ReaGAN treats each data point (node) as an autonomous agent that can plan, remember, and actively retrieve information from the entire network. This helps build a much smarter, more complete understanding of the data's global relationships.
Coolest part? It achieves impressive results without needing costly retraining of its core AI model!
#AI #DataScience #GraphLearning #GNN #LLM #MachineLearning
Read the paper: arxiv.org/abs/2508.00429
2
18

12 Jul 2025
Heading to #ICML2025 in Vancouver next week! Thrilled to present:
🖥️ UI-Vision: A Desktop-centric GUI Benchmark for Visual Perception and Interaction @ Main Conference
📆 Mon, July 15 | 🕑 2:00–4:30 PM
📍 West Hall B2–B3 | Poster W-400
We introduce a large-scale benchmark to evaluate GUI agents on perception, grounding, and interaction across 83 real-world desktop platforms.
🖼️ Paper2Poster: Towards Multimodal Poster Automation from Scientific Papers @ MAS Workshop (Oral)
📆 Thu, July 18 | 🕗 8:30 AM (Time TBD)
📍 Room 109–110
We propose a multi-agent pipeline (PosterAgent) to generate .pptx posters from full papers, evaluated on visual alignment, coherence, and a novel comprehension test.
Grateful to collaborate with @real_weipang, @PShravannayak, @KevinQHLin , Philip Torr & the amazing team @ServiceNowRSRCH.
If you're interested in agents, multimodal reasoning, graph learning and multimodal data management, let’s connect!
#ICML #MAS2025 #LLMs #VLMs #GUIAgents #GraphLearning #PosterGeneration #GenerativeAI
1
15
290

We welcome you to submit your papers and participate in CSoNet 2025. csonet-conf.github.io
#CFP #Conference #papers #SocialNetworks #BigData #NetworkScience #AI #GraphLearning

2
158

29 May 2025
#CallforPaper
💫 Advances in Graph Learning and Representation Models for Complex Network Analysis
This SI aims to bring together leading-edge research that explores the design, implementation, and application of #GraphLearning and #RepresentationModel.
mdpi.com/journal/BDCC/specia…


2
51

28 May 2025
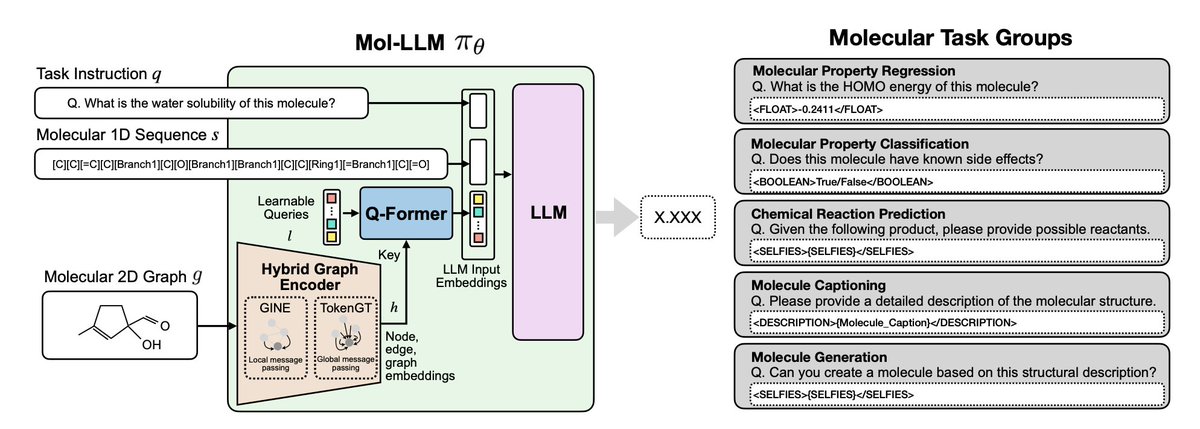
Mol-LLM: Multimodal Generalist Molecular LLM with Improved Graph Utilization
1.Mol-LLM is the first generalist molecular language model that robustly integrates both 1D molecular sequences and 2D graph structures across a broad range of tasks, including property prediction, reaction prediction, and molecule generation.
2.At the core of its innovation is MolPO (Molecular structure Preference Optimization), a multimodal fine-tuning strategy that teaches the model to distinguish between correct and perturbed molecular graphs—enhancing structure utilization without additional supervision.
3.Unlike prior multimodal models that rely on naive next-token prediction, Mol-LLM uses MolPO to optimize structural preferences, significantly boosting performance and generalization, especially on out-of-distribution benchmarks.
4.To maximize graph expressivity, Mol-LLM combines two GNN backbones—GINE and TokenGT—into a hybrid encoder, with pre-training on both functional group prediction and SELFIES reconstruction for rich molecular representation.
5.A Q-Former module bridges the molecular graph and language input, projecting concatenated graph embeddings into a fixed-length token sequence compatible with a Mistral-7B backbone, which is extended with domain-specific tokens for regression, classification, and generation tasks.
6.Mol-LLM is trained on a 3.3M instance multimodal instruction-tuning dataset covering five task groups, constructed from sources such as SMolInstruct, Mol-Instructions, and ChEBI-20, enabling flexible handling of diverse molecular queries.
7.Across MoleculeNet and other molecular benchmarks, Mol-LLM outperforms state-of-the-art generalist models like LlaSMol, ChemDFM, and 3D-MoLM, notably in reaction prediction and property regression where structural information is key.
8.Ablation studies reveal that the addition of MolPO improves both performance and graph discrimination ability across all tasks. GNN pre-training alone also yields consistent gains, especially on tasks requiring recognition of global molecular features.
9.Even in text-only settings (e.g., molecule generation or captioning), Mol-LLM matches or exceeds baselines, and its ability to leverage graphs does not impair performance when structural input is absent—highlighting its multimodal flexibility.
10.Evaluation on out-of-distribution datasets (AqSol and ORDerly) confirms Mol-LLM’s strong generalization, a result attributed to the combination of graph-aware training and broad instruction coverage.
11.Mol-LLM establishes a new paradigm in molecular LLMs by unifying instruction tuning with graph-aware preference optimization, laying the foundation for future generalist systems in chemical and biomedical domains.
💻Code: github.com/LGAI-Research/Mol…
📜Paper: arxiv.org/abs/2502.02810
#MolLLM #GraphLearning #DrugDiscovery #LLM #MolecularAI #ChemistryAI #MultimodalAI
1
7
621
23 May 2025
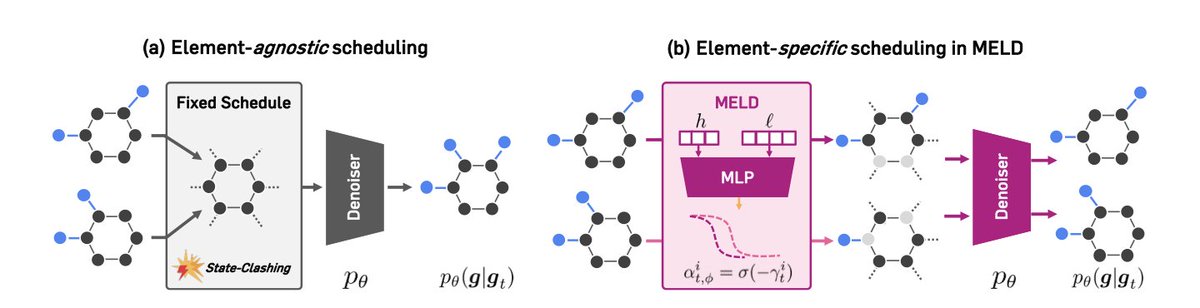
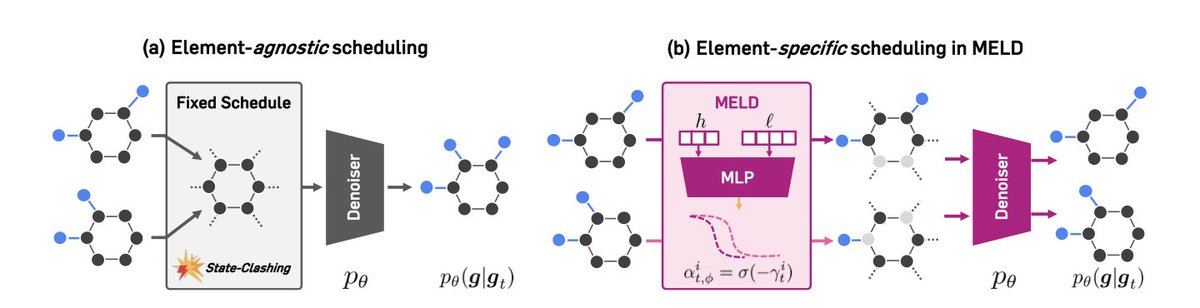
Learning Flexible Forward Trajectories for Masked Molecular Diffusion
1.This paper introduces MELD, a novel masked diffusion model for molecular graph generation that learns element-wise corruption schedules. By avoiding the collapse of distinct molecular graphs into indistinguishable masked states, MELD dramatically improves validity and property alignment in molecule generation tasks.
2.Standard masked diffusion models (MDMs) apply uniform noise across graph elements, which causes a “state-clashing” problem where different molecules converge to similar intermediate representations. MELD addresses this with per-atom and per-bond noise scheduling.
3.The core innovation in MELD is a learnable noise scheduling network that outputs distinct corruption rates for each node and edge, dynamically controlling which molecular components are masked at each timestep to avoid trajectory collisions.
4.Unlike autoregressive masked diffusion models (e.g., GraphARM), MELD preserves parallelism and achieves higher chemical validity. For example, it boosts validity on ZINC250K from 15% (baseline MDM) to 93%, a more than 6-fold improvement.
5.In unconditional generation on QM9 and ZINC250K, MELD achieves state-of-the-art performance across key metrics including Fréchet ChemNet Distance (FCD), NSPDK, uniqueness, and scaffold similarity—outperforming DiGress, GraphDF, and continuous diffusion models like GDSS and EDP-GNN.
6.In property-conditioned generation (Polymer dataset), MELD surpasses previous best methods like GraphDiT and MOOD, reducing mean absolute error in property alignment by up to 16% and outperforming across synthesizability and gas permeability targets.
7.Extensive ablation studies confirm that element-wise scheduling is crucial: MELD outperforms both class-wise and unified (fixed) noise scheduling, especially in avoiding chemically implausible structures at late diffusion steps.
8.Visual analyses reveal MELD’s denoising strategy prioritizes nodes before edges and functional groups before frameworks, reconstructing valid molecular graphs earlier in the reverse process than baselines with fixed scheduling.
9.MELD’s forward noise schedule is learned using a parameterized MLP, and optimized with a straight-through Gumbel-Softmax trick, allowing gradient flow through discrete sampling operations without sacrificing differentiability.
10.This work positions MELD as a robust and generalizable framework for molecule generation, with clear implications for improving sample quality, controllability, and downstream optimization in drug discovery and materials design.
📜Paper: arxiv.org/abs/2505.16790
#MolecularGeneration #DiffusionModels #GraphLearning #ComputationalChemistry #MaskedDiffusion #MELD #GenerativeModels #DrugDiscovery #ChemicalSpace #AI4Science
2
6
696
23 May 2025
Learning Flexible Forward Trajectories for Masked Molecular Diffusion
1.This paper introduces MELD, a novel masked diffusion model for molecular graph generation that learns element-wise corruption schedules. By avoiding the collapse of distinct molecular graphs into indistinguishable masked states, MELD dramatically improves validity and property alignment in molecule generation tasks.
2.Standard masked diffusion models (MDMs) apply uniform noise across graph elements, which causes a “state-clashing” problem where different molecules converge to similar intermediate representations. MELD addresses this with per-atom and per-bond noise scheduling.
3.The core innovation in MELD is a learnable noise scheduling network that outputs distinct corruption rates for each node and edge, dynamically controlling which molecular components are masked at each timestep to avoid trajectory collisions.
4.Unlike autoregressive masked diffusion models (e.g., GraphARM), MELD preserves parallelism and achieves higher chemical validity. For example, it boosts validity on ZINC250K from 15% (baseline MDM) to 93%, a more than 6-fold improvement.
5.In unconditional generation on QM9 and ZINC250K, MELD achieves state-of-the-art performance across key metrics including Fréchet ChemNet Distance (FCD), NSPDK, uniqueness, and scaffold similarity—outperforming DiGress, GraphDF, and continuous diffusion models like GDSS and EDP-GNN.
6.In property-conditioned generation (Polymer dataset), MELD surpasses previous best methods like GraphDiT and MOOD, reducing mean absolute error in property alignment by up to 16% and outperforming across synthesizability and gas permeability targets.
7.Extensive ablation studies confirm that element-wise scheduling is crucial: MELD outperforms both class-wise and unified (fixed) noise scheduling, especially in avoiding chemically implausible structures at late diffusion steps.
8.Visual analyses reveal MELD’s denoising strategy prioritizes nodes before edges and functional groups before frameworks, reconstructing valid molecular graphs earlier in the reverse process than baselines with fixed scheduling.
9.MELD’s forward noise schedule is learned using a parameterized MLP, and optimized with a straight-through Gumbel-Softmax trick, allowing gradient flow through discrete sampling operations without sacrificing differentiability.
10.This work positions MELD as a robust and generalizable framework for molecule generation, with clear implications for improving sample quality, controllability, and downstream optimization in drug discovery and materials design.
📜Paper: arxiv.org/abs/2505.16790
#MolecularGeneration #DiffusionModels #GraphLearning #ComputationalChemistry #MaskedDiffusion #MELD #GenerativeModels #DrugDiscovery #ChemicalSpace #AI4Science
4
16
1,037
19 May 2025
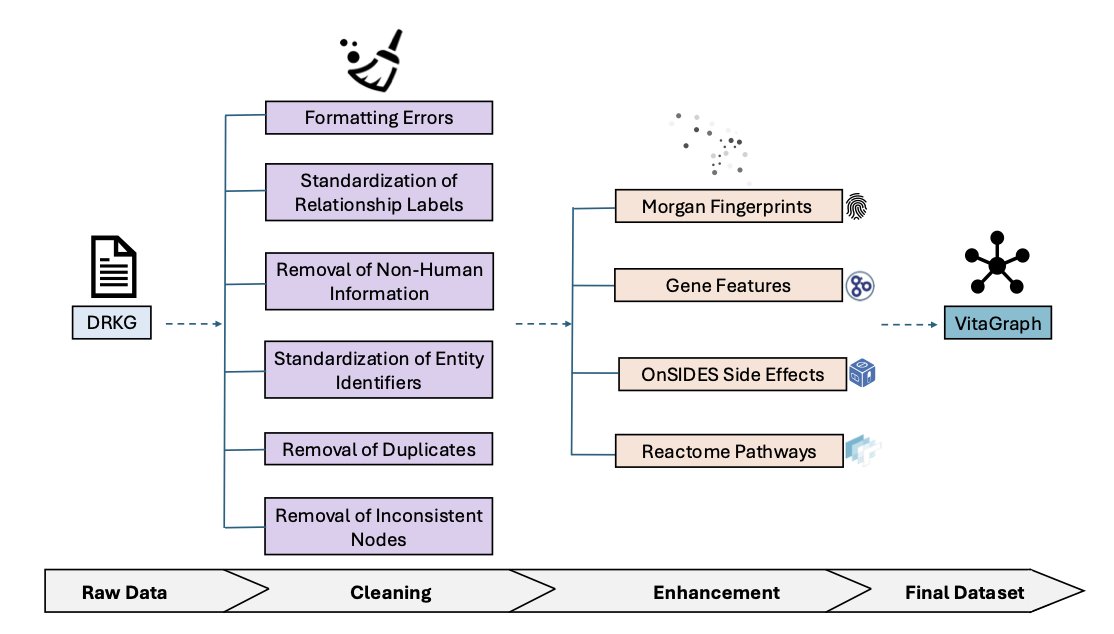
VITAGRAPH: Building a Knowledge Graph for Biologically Relevant Learning Tasks
1.VITAGRAPH is a new biological knowledge graph purpose-built for machine learning tasks in network medicine, including drug repurposing, protein–protein interaction (PPI) prediction, and side effect identification.
2.The graph is constructed by extensively cleaning and enriching the popular Drug Repurposing Knowledge Graph (DRKG), removing redundancy, non-human data, inconsistent labels, and unresolvable identifiers.
3.VITAGRAPH integrates biologically meaningful node features like molecular fingerprints (for compounds) and Gene Ontology-derived binary vectors (for genes), enabling more expressive graph learning.
4.The authors enhanced DRKG by reintroducing pathway nodes via Reactome, and incorporating high-confidence compound–side effect edges from OnSIDES, adding 2,153 pathways and over 339,000 new edges.
5.They excluded compounds without valid SMILES representations and used Morgan fingerprints (radius 2, 2048 bits) for standardized chemical features, improving chemical interpretability and utility.
6.Gene features were collapsed from star-shaped subgraphs into one-hot encoded vectors describing pathways, molecular functions, biological processes, and cellular components—yielding biologically rich node representations.
7.The final VITAGRAPH consists of 48,058 nodes and 4,004,583 edges, after eliminating nearly 2 million duplicate or inconsistent rows from DRKG.
8.Benchmarking across three tasks (drug repurposing, PPI prediction, and side-effect detection) using R-GCN and CompGCN shows that VITAGRAPH matches or outperforms DRKG—even when DRKG's results were inflated due to data leakage.
9.Detailed leakage analysis reveals that DRKG suffers from up to 65% interaction overlap between training and test sets in the PPI and drug repurposing tasks, undermining its benchmarking reliability.
10.In contrast, VITAGRAPH avoids such leakage, providing a clean and biologically relevant foundation for graph machine learning and link prediction experiments.
11.VITAGRAPH’s modular pipeline is publicly available, letting users reproduce, customize, or extend the graph to suit specific biomedical questions or integrate new datasets over time.
12.By releasing both the dataset and code, the authors establish VITAGRAPH as a reproducible and high-quality benchmark to support future advances in computational biology and biomedical AI.
💻Code: github.com/GiDeCarlo/VitaGra…
📜Paper: arxiv.org/abs/2505.11185v1
#KnowledgeGraph #Bioinformatics #ComputationalBiology #DrugRepurposing #PPI #GNN #GraphLearning #BiomedicalAI #DRKG #VitaGraph #MachineLearning
2
10
943