
Jun 3
Ph.D. Opportunities in Molecular Machine Learning & Generative AI for Organic Chemistry
📍 Location: IIT Bombay, India
This is a posting intended for prospective students who are considering pursuing a Ph.D. in advanced areas of molecular machine learning and generative AI for organic chemistry.
We have a couple of positions open in the July 2026 admission cycle for which the admission offers were already sent out. What preparedness is required for this position? -Willingness to learn! Everything else would follow.
Check our website for more details on publications (chem.iitb.ac.in/~sunoj/publi…) and to check the career profiles of past students from the lab (chem.iitb.ac.in/~sunoj/alumn…).
#Phd #vacancy #phdposition #phdstudent #research @jobRxiv @iitbombay @Chemistry_IITB #Chemistry
#PhD #PhDOpportunity #PhDPosition #MachineLearning #GenerativeAI #MolecularMachineLearning #OrganicChemistry #IITBombay #Chemistry #ResearchOpportunity #AIforScience #ComputationalChemistry
5
24
2,099
SynCraft: An Integrated Web Server for ADMET-Aware Retrosynthesis and Molecular Design
1. SynCraft is a web server that unifies multi-step retrosynthetic planning with real-time ADMET evaluation, so every intermediate generated during route search is assessed for safety and drug-likeness without exporting molecules to separate tools.
2. The key practical feature is automatic, color-coded flagging of potentially hazardous intermediates during route exploration (e.g., genotoxicity-related alerts), addressing a common blind spot where conventional CASP tools optimize feasibility but ignore intermediate toxicity.
3. In an imatinib case study, SynCraft flags a well-known mutagenic intermediate, 4-methyl-1,2-phenylenediamine (ICH M7 Class 2), immediately when it appears in the tree (ClinTox 100%, ToxCast 0.18), while standard retrosynthesis outputs can present the same routes with no toxicity warning.
4. The retrosynthesis engine is template-based and deterministic: BFS search with InChIKey cycle detection, depth capped at 6 steps (chosen because 94.3% of molecules in a calibration set were solvable within 6), and termination when reaching a 22.4M commercial building-block database (ZINC15 eMolecules).
5. SynCraft uses a large reaction template library (384,512 templates) derived from USPTO-MIT plus Retro* templates, interleaved by frequency and weighted with an exponential decay scheme to balance solve rate vs runtime and reduce instability from rare templates.
6. Route ranking is multi-objective, combining synthetic accessibility (SA), synthetic complexity (SC), worst-intermediate ADMET signal, and step count via a Route Score; weights were tuned against chemist preference ratings on a held-out set (reported Spearman ρ = 0.71).
7. ADMET prediction is powered by MolMVC, a multi-view contrastive learning framework combining 1D (Transformer on fingerprints), 2D (GIN Graph Transformer), and 3D (SchNet-based Graph Transformer on ETKDG/MMFF94 conformers), enabling intermediate-level predictions beyond simple 2D-only representations.
8. Safety flagging uses calibrated operational thresholds (e.g., ClinTox > 0.70; ToxCast < 0.40), motivated by ROC analysis and external validation; SynCraft also reports Lipinski compliance and QED for intermediates, plus SA (TwistDAN) and SCScore for synthesis difficulty context.
9. Benchmark and workflow results emphasize integration rather than higher solve rate: retrosynthesis performance is comparable to AiZynthFinder, but SynCraft evaluates all intermediates across all routes and reduces manual steps; a timing study reports ~84–114 s end-to-end vs ~1,230–1,410 s for a sequential retrosynthesis manual ADMET workflow (>91% reduction driven by eliminating per-intermediate manual evaluation and file handling).
💻Code: github.com/Q-Aljanabi/SynCra…
📜Paper: doi.org/10.1093/nar/gkag463
#Retrosynthesis #ADMET #Cheminformatics #DrugDiscovery #ToxicityPrediction #MolecularMachineLearning #WebServer #ComputationalChemistry #MedicinalChemistry

6
22
1,929

26 Nov 2025
Open (computer science) PhD positions in #MachineLearning for #MaterialsScience and #Chemistry:
pse.kit.edu/english/karriere…
pse.kit.edu/english/karriere…
pse.kit.edu/english/karriere…
Please share in your networks!
#AI4Science #MaterialsDiscovery #MolecularMachineLearning #SelfDrivingLabs

3
3
480

22 Oct 2025
DeepChem Equivariant: SE(3)-Equivariant Support in an Open-Source Molecular Machine Learning Library
1. The integration of SE(3)-equivariant neural networks into the DeepChem library represents a significant leap forward in molecular machine learning. These networks ensure that model outputs transform predictably with input coordinate changes, crucial for tasks like molecular property prediction and protein structure modeling.
2. The core innovation lies in the implementation of SE(3)-equivariant models such as SE(3)-Transformer and Tensor Field Networks within DeepChem. This modular infrastructure broadens the application of equivariant methods, making them accessible to scientists with minimal deep learning background.
3. The framework efficiently computes equivariant features using spherical harmonics and irreducible representations. This approach not only respects 3D geometric symmetries but also offers a practical and well-documented suite of tools for equivariant molecular modeling.
4. The implementation includes complete training pipelines and a toolkit of equivariant utilities, supported with comprehensive tests and documentation. This ensures robustness and fosters contributions from the community, making it easier to build, train, and evaluate models.
5. Experiments on the QM9 dataset demonstrate that DeepChem's SE(3)-equivariant models achieve comparable performance to state-of-the-art methods, highlighting the effectiveness of incorporating attention mechanisms into roto-translation-equivariant models.
6. Future work includes implementing LieConv in DeepChem-Equivariant to improve efficiency, particularly in basis construction. Caching precomputed bases in DeepChem's GraphData class is also proposed to avoid bottlenecks during training and improve scalability.
📜Paper: arxiv.org/abs/2510.16897
#DeepChem #EquivariantNeuralNetworks #MolecularMachineLearning #SE3Equivariance #OpenSource #MolecularModeling #DrugDiscovery #MaterialsScience

4
21
1,122
29 Aug 2025
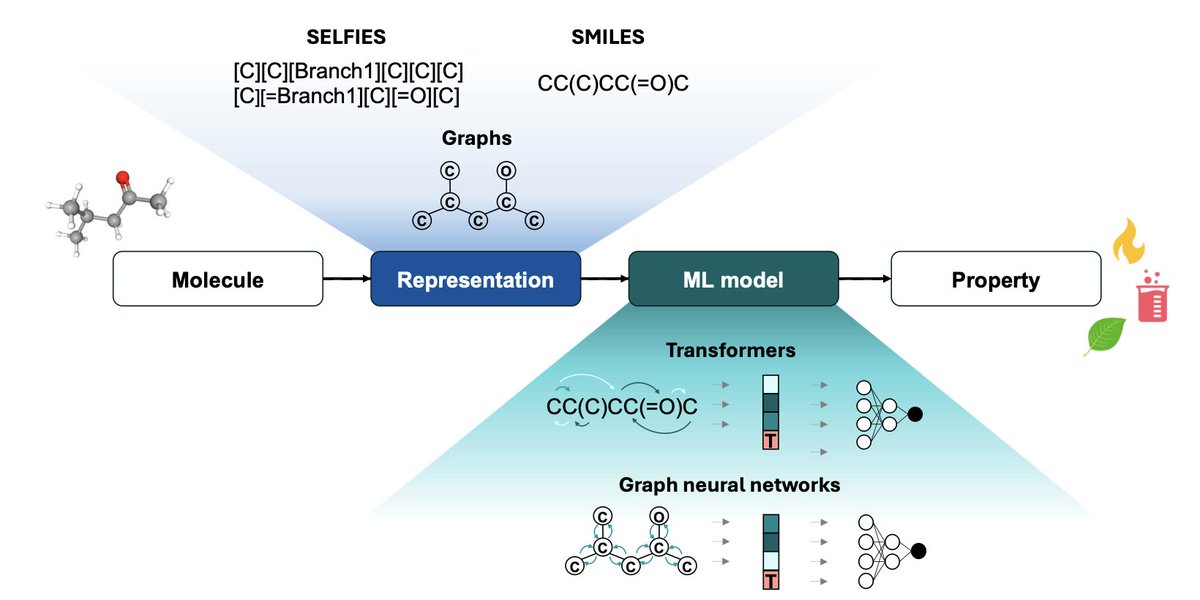
Molecular Machine Learning in Chemical Process Design
1. This perspective article explores the potential of molecular machine learning (ML) in chemical process engineering, highlighting its ability to predict molecular properties and design new molecules with high accuracy. The integration of ML into chemical process design could significantly accelerate the identification of novel molecules and processes.
2. The article reviews state-of-the-art molecular ML models, including graph neural networks (GNNs) and transformers, which have shown remarkable performance in predicting properties of both pure components and mixtures. These models often outperform traditional methods in chemical engineering, such as UNIFAC and COSMO-RS.
3. A key innovation discussed is the incorporation of physicochemical knowledge into ML models in a hybrid or physics-informed manner. This approach not only enhances the accuracy of predictions but also ensures thermodynamic consistency, which is crucial for practical applications.
4. The authors advocate for leveraging molecular ML at the chemical process scale, which remains largely unexplored. They propose integrating ML models into process design and optimization formulations, suggesting that this could lead to more sustainable and efficient chemical processes.
5. The article emphasizes the importance of creating benchmarks and validating proposed molecular candidates in collaboration with the chemical industry. This would help in establishing practical standards and ensuring the reliability of ML-driven molecular design.
6. The authors also highlight the potential of generative ML models for computer-aided molecular design (CAMD), which can explore the chemical space beyond traditional methods. This opens up new possibilities for discovering molecules with desired properties.
7. The article concludes by calling for strong collaboration between academia and industry to improve the development and reliability of molecular ML models. This collaboration is seen as essential for advancing the practical application of these models in industrial settings.
📜Paper: arxiv.org/abs/2508.20527v1
#MolecularMachineLearning #ChemicalProcessDesign #GraphNeuralNetworks #Transformers #AIinChemistry #SustainableChemistry
2
4
18
1,575

29 Aug 2025
Excellence at the intersection of data science, cheminformatics & organic #chemistry - Congrats Florian on a poster award! @GloriusGroup, we use AI, HTE & HPC to advance catalysis & materials. 💻😀⚗️ #Cheminformatics #DataScience @spp2363
#MolecularMachineLearning @uni_muenster


5
41
16,116
28 Aug 2025
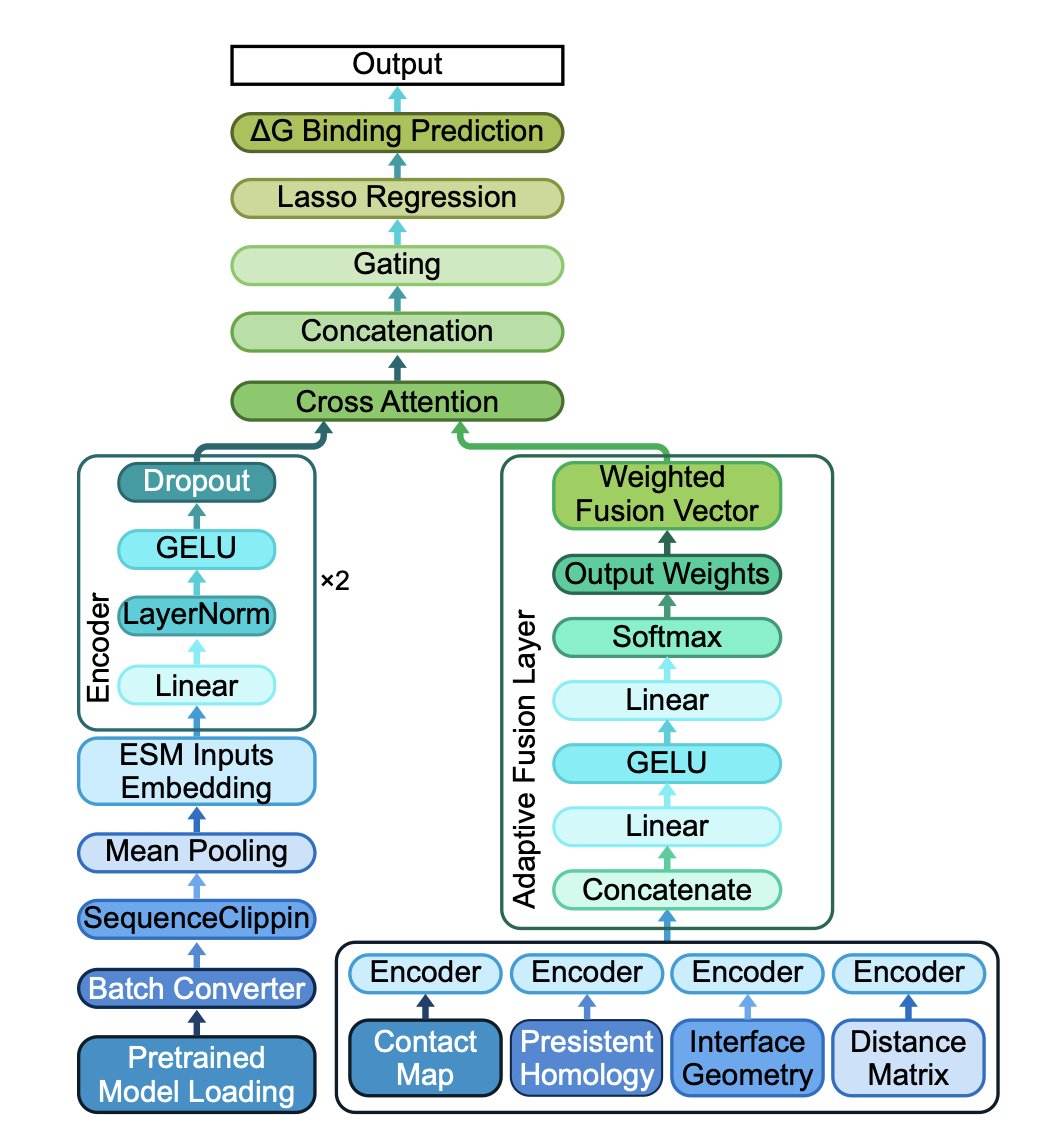
TOPOBIND: MULTI-MODAL PREDICTION OF ANTIBODY-ANTIGEN BINDING FREE ENERGY VIA SEQUENCE EMBEDDINGS AND STRUCTURAL TOPOLOGY
1. A novel framework called TopoBind integrates sequence-based representations from pre-trained protein language models (ESM-2) with a set of topological features to predict antibody-antigen binding free energy, achieving state-of-the-art accuracy in binding free energy prediction.
2. TopoBind extracts contact map metrics, interface geometry descriptors, distance map statistics, and persistent homology invariants to capture both local and global structural organization within individual proteins and across the antibody-antigen interface.
3. The model employs a cross-attention mechanism to fuse diverse modalities effectively, enhancing the prediction of binding free energy. It also uses an adaptive feature fusion mechanism to dynamically weight different topological submodules, improving model generalization.
4. Experiments on a curated dataset of 303 antibody-antigen complexes show that TopoBind consistently outperforms sequence-only and conventional structural models in both regression and classification settings.
5. Ablation studies demonstrate the importance of each architectural component, including the adaptive fusion module and sparse linear modeling, in enhancing the model's generalization ability.
6. The study also explores the sensitivity of topological parameters, finding that the model performs best with specific settings for the interface contact distance threshold and the number of top-k persistent homology lifetimes retained.
📜Paper: arxiv.org/abs/2508.19632
#TopoBind #AntibodyAntigenBinding #ProteinLanguageModels #TopologicalDataAnalysis #StructuralBioinformatics #CrossAttention #ProteinRepresentationLearning #MolecularMachineLearning
2
22
1,402
13 Aug 2025
Molecular deep learning at the edge of chemical space
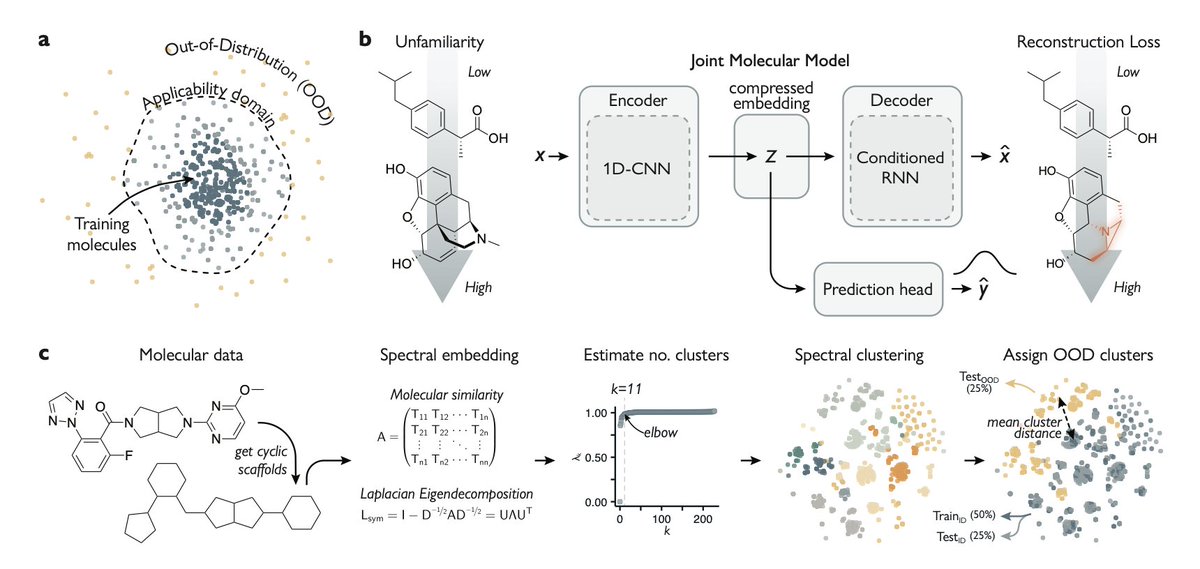
1. This study introduces a novel approach in molecular machine learning that combines molecular property prediction with molecular reconstruction to enhance the model's ability to generalize beyond its training data. The key innovation is the introduction of a metric called "unfamiliarity" which quantifies how much a molecule deviates from the training distribution.
2. The authors demonstrate that the unfamiliarity metric effectively identifies out-of-distribution molecules and serves as a reliable predictor of classifier performance. This is particularly significant in the context of drug discovery, where identifying structurally novel bioactive molecules is crucial.
3. The study spans over 30 bioactivity datasets, showing that unfamiliarity not only detects distribution shifts but also correlates strongly with classifier performance. This suggests that unfamiliarity can guide the discovery of diverse and structurally novel molecules, which is a major challenge in early drug discovery.
4. The authors experimentally validated their approach by screening molecules for two clinically relevant kinases, discovering several compounds with low micromolar potency and limited similarity to training molecules. This highlights the practical applicability of the unfamiliarity metric in wet lab settings.
5. The Joint Molecular Model (JMM) used in this study incorporates a semi-supervised autoencoder architecture, allowing simultaneous training for molecular reconstruction and property prediction. This joint learning approach breaks new ground in leveraging reconstruction capabilities for out-of-distribution estimation.
6. The study also explores the use of unfamiliarity in large-scale virtual screening campaigns, demonstrating its ability to detect strong distribution shifts that traditional metrics might miss. This suggests that unfamiliarity can be a valuable tool for navigating chemical space in virtual screening scenarios.
7. The authors propose that unfamiliarity can complement existing concepts like the applicability domain and uncertainty estimation, providing a more holistic approach to estimating prediction reliability in molecular machine learning.
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularMachineLearning #DeepLearning #DrugDiscovery #ChemicalSpace #NoveltyDetection
1
6
39
2,316
23 Jun 2025
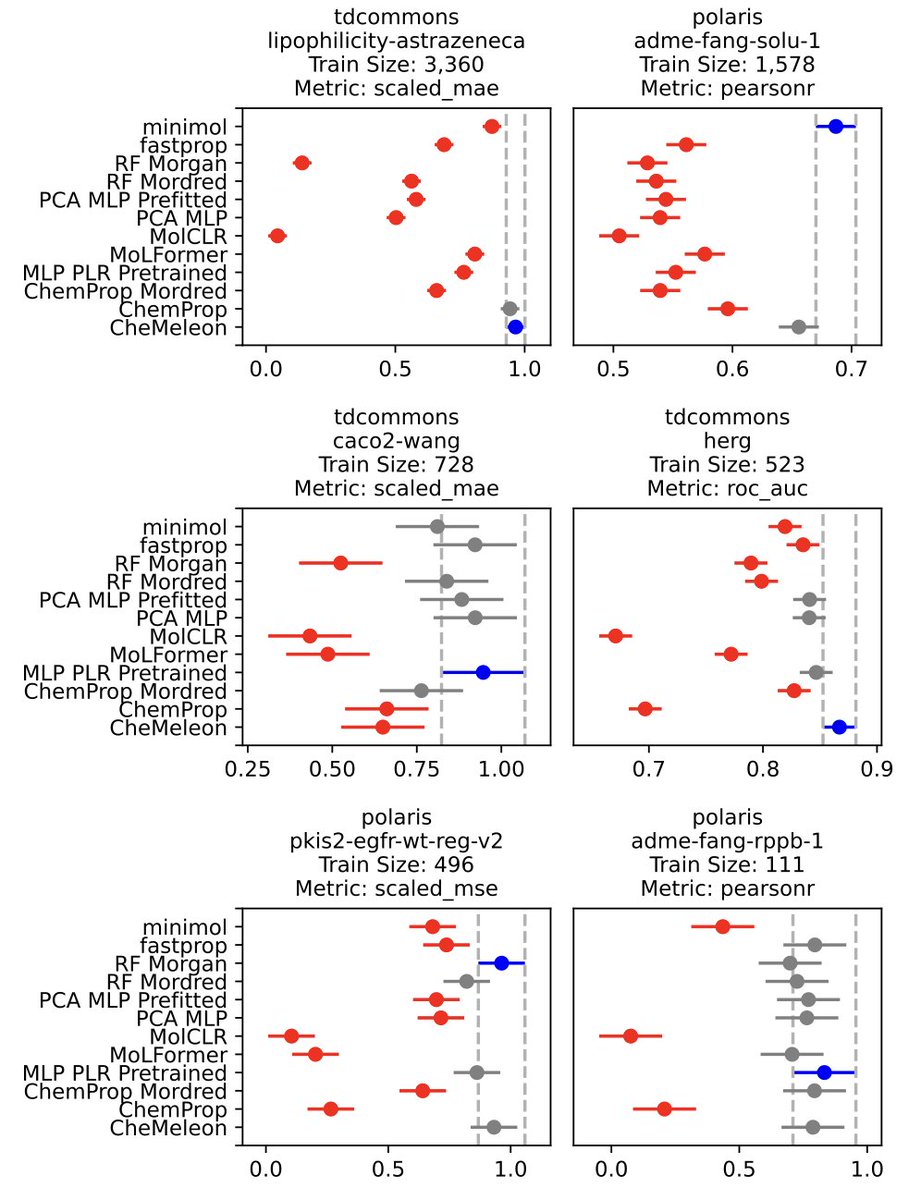
Descriptor-based Foundation Models for Molecular Property Prediction
1.CheMeleon is a new foundation model for molecular property prediction, pre-trained on deterministic molecular descriptors instead of noisy experimental or quantum mechanical data. It uses a Directed Message-Passing Neural Network (D-MPNN) to predict Mordred descriptors in a noise-free setting.
2.By pretraining on low-noise descriptors rather than stochastic experimental or biased QM simulations, CheMeleon learns rich molecular representations that transfer well to diverse downstream tasks. This enables better generalization, especially on small datasets.
3.On 58 benchmark datasets, CheMeleon achieves a 79% win rate on Polaris tasks and a 97% win rate on MoleculeACE benchmarks—outperforming Random Forest, Chemprop, and several recent foundation models like MoLFormer and minimol.
4.CheMeleon is especially effective in low-data regimes, a common weakness for learned representation models like Chemprop. Pretraining on descriptors allows the model to avoid the need for large datasets to simultaneously learn features and mappings.
5.In contrast to other foundation models that use experimental or QM properties for supervision, CheMeleon’s labels (Mordred descriptors) are deterministic, cheap to compute, and scalable—enabling pretraining on over 1 million molecules from PubChem.
6.Despite its strong overall performance, CheMeleon—like all tested models—struggles to resolve activity cliffs (subtle structural changes with large functional impact), highlighting a persistent challenge in molecular machine learning.
7.t-SNE projections of CheMeleon embeddings show that the model captures subtle chemical features and organizes molecules by structural similarity. This makes its learned representation interpretable and potentially more useful in medicinal chemistry.
8.CheMeleon’s success suggests that simple, hand-engineered molecular descriptors, long considered obsolete, can be used as powerful pretraining targets for modern deep learning architectures when combined with foundation model training.
9.The authors suggest that alternative descriptor sets (e.g., DRAGON, PaDEL) or different unlabeled datasets (e.g., ChEMBL, COCONUT) could be explored to tailor pretraining for specific chemical domains.
10.CheMeleon is open-source, permissively licensed, and fully integrated with the Chemprop framework, making it easy for practitioners to adopt and extend.
💻Code:
github.com/JacksonBurns/CheM…
📜Paper:
arxiv.org/abs/2506.15792v1
#Cheminformatics #MolecularMachineLearning #FoundationModels #DrugDiscovery #DeepLearning #GraphNeuralNetworks
5
18
959

6 May 2025
Join the Glorius Group at the intersection of cheminformatics, data science & synthetic #chemistry! We use AI, HTE & HPC to advance catalysis & materials. PhD/Postdoc opportunities available! 💻😀⚗️ #Cheminformatics #DataScience @spp2363 #MolecularMachineLearning @uni_muenster

8
87
7,034
1 Mar 2025
Neural Graph Matching Improves Retrieval Augmented Generation in Molecular Machine Learning
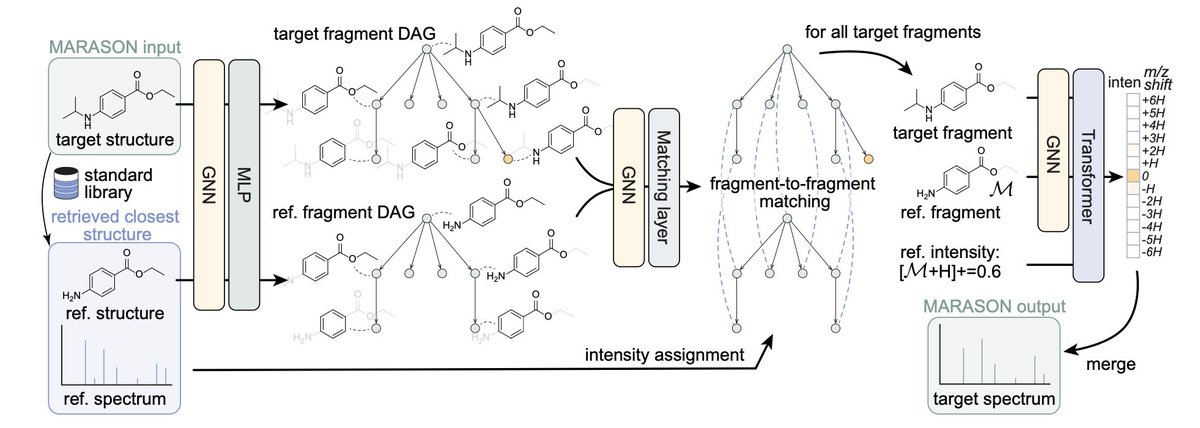
1/ This study introduces MARASON, a novel approach that combines neural graph matching with retrieval-augmented generation (RAG) to improve mass spectrum simulation accuracy in molecular machine learning.
2/ MARASON enhances traditional molecular models by using a neural graph matching technique that learns affinities between node and edge pairs in molecular graphs, ensuring more accurate and robust structural alignment of retrieved molecules to the query structure.
3/ The integration of neural graph matching into the RAG framework allows MARASON to better predict mass spectra by aligning molecular fragments more accurately, improving spectrum intensity predictions in mass spectrometry simulations.
4/ The model outperforms traditional graph matching methods, which rely on predefined affinity metrics, by offering a learnable, end-to-end approach. This flexibility allows MARASON to handle complex, noisy real-world molecular data more effectively.
5/ MARASON achieves a top-1 retrieval accuracy of 28%, a significant improvement over the non-RAG baseline of 19%, and outperforms other retrieval-augmented methods, demonstrating its power in simulating mass spectra with high accuracy.
6/ The paper shows that MARASON's neural graph matching approach surpasses both naive RAG models and traditional graph matching methods, making it a new state-of-the-art method for mass spectrum simulation in molecular machine learning.
7/ MARASON’s ability to match molecular fragments based on fragmentation DAGs highlights its potential for real-world applications in chemical and biological fields, improving the accuracy of molecular discovery and structural elucidation tasks.
8/ The results suggest that neural graph matching-based RAG is a promising direction for enhancing molecular machine learning tasks, such as structure-property prediction, beyond mass spectrum simulation.
📜Paper: arxiv.org/abs/2502.17874
#AI #MolecularMachineLearning #GraphMatching #MassSpectrometry #MolecularModeling #MachineLearning #ComputationalChemistry #Bioinformatics
1
2
8
2,164
1 Mar 2025
Neural Graph Matching Improves Retrieval Augmented Generation in Molecular Machine Learning
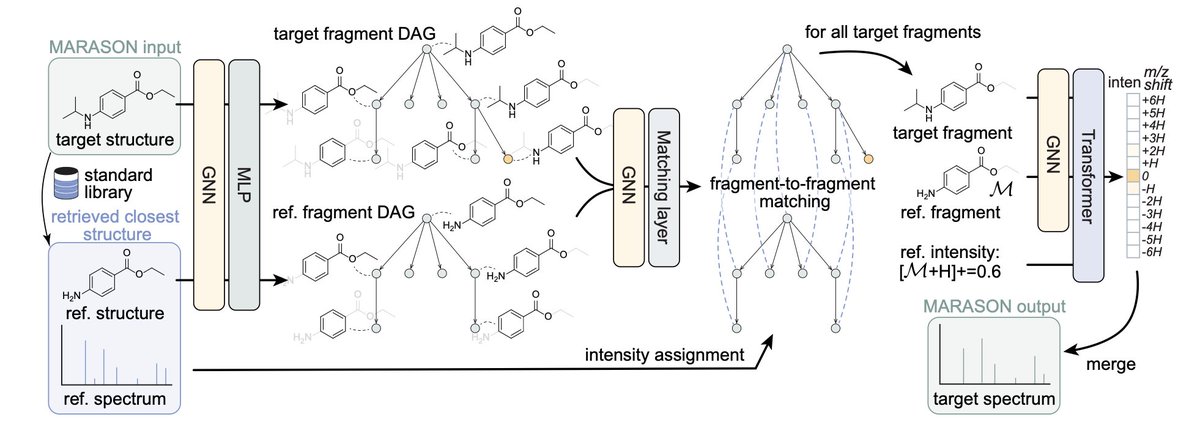
1/ This study introduces MARASON, a novel approach that combines neural graph matching with retrieval-augmented generation (RAG) to improve mass spectrum simulation accuracy in molecular machine learning.
2/ MARASON enhances traditional molecular models by using a neural graph matching technique that learns affinities between node and edge pairs in molecular graphs, ensuring more accurate and robust structural alignment of retrieved molecules to the query structure.
3/ The integration of neural graph matching into the RAG framework allows MARASON to better predict mass spectra by aligning molecular fragments more accurately, improving spectrum intensity predictions in mass spectrometry simulations.
4/ The model outperforms traditional graph matching methods, which rely on predefined affinity metrics, by offering a learnable, end-to-end approach. This flexibility allows MARASON to handle complex, noisy real-world molecular data more effectively.
5/ MARASON achieves a top-1 retrieval accuracy of 28%, a significant improvement over the non-RAG baseline of 19%, and outperforms other retrieval-augmented methods, demonstrating its power in simulating mass spectra with high accuracy.
6/ The paper shows that MARASON's neural graph matching approach surpasses both naive RAG models and traditional graph matching methods, making it a new state-of-the-art method for mass spectrum simulation in molecular machine learning.
7/ MARASON’s ability to match molecular fragments based on fragmentation DAGs highlights its potential for real-world applications in chemical and biological fields, improving the accuracy of molecular discovery and structural elucidation tasks.
8/ The results suggest that neural graph matching-based RAG is a promising direction for enhancing molecular machine learning tasks, such as structure-property prediction, beyond mass spectrum simulation.
📜Paper: arxiv.org/abs/2502.17874
#AI #MolecularMachineLearning #GraphMatching #MassSpectrometry #MolecularModeling #MachineLearning #ComputationalChemistry #Bioinformatics
1
3
10
1,727

6 Jun 2024
Molecular Nano Neural Networks (M3N):In-Body Intelligence for the IoBNT
#NanoCyberInterface
#InternetofBioNanoThings
#NeuralEngineering
#MolecularMachineLearning
#MolecularEngineering
techrxiv.org/users/689760/ar…

1
1
34
31 May 2024
Molecular Nano Neural Networks (M3N):In-Body Intelligence for the IoBNT
#InternetofBioNanoThings
#MolecularMachineLearning
#MolecularEngineering
'Bio-Nano Things (BNTs) used in the human body...' techrxiv.org/users/689760/ar…

6
6
239
30 May 2024
Micro-Body-Area-Network
#WirelessIntraBodyNanoCommunication
#NanoCyberInterface
#NanoNodes
#MolecularEngineering
#MolecularMachineLearning
IEEE 802.15.4
IEEE 802.15.6
Human PHY layer: narrowband, ultra-wideband & HUMAN BODY COMMUNICATION
future-forum.org.cn/dl/20112…


1
21
20
1,373
30 May 2024
Molecular Nano Neural Networks (M3N):In-Body Intelligence for the IoBNT
#NanoCyberInterface
#PANACEA
#InternetofBioNanoThings #MolecularMachineLearning #NeuralEngineering
Angerbauer
techrxiv.org/users/689760/ar…
Novel Nano-Scale Computing Unit for the IoBNT
researchgate.net/publication…

30 May 2024

How do we connect the Human Body to the Internet...?
Molecular Nano Neural Networks (M3N): In-Body Intelligence for the Internet of Bio-Nano Things
#NanoCyberInterface
#MolecularEngineering
#IoBNT
#MolecularMachineLearning
Stefan Angerbauer
researchgate.net/publication…
19
25
2,060
30 May 2024
How do we connect the Human Body to the Internet...?
Molecular Nano Neural Networks (M3N): In-Body Intelligence for the Internet of Bio-Nano Things
#NanoCyberInterface
#MolecularEngineering
#IoBNT
#MolecularMachineLearning
Stefan Angerbauer
researchgate.net/publication…
30 May 2024
Internet of Bio-Nano Things Nanoscale Computing through the Human Body
Machine Learning in the IoBNT
#MolecularEngineering
#NeuralEngineering
Novel Nano-Scale Computing Unit for the IoBNT: Concept and Practical Considerations
tkn.tu-berlin.de/team/torres…
researchgate.net/publication…


7
63
71
8,929
29 May 2024
Yo Grok what is the role of the Bio-Cyber Interface Human Physical layer in 6G?
#NanoNodes #THz within the HUMAN BODY
#IntraBodyNanoNetworks
in VIVO Wireless NANO SENOR NETWORKS
#InternetofBioNanoThings #IoBNT
#MedicalBodyAreaNetwork
#MolecularMachineLearning
IEEE 802.15.4

1
44
38
1,769
29 May 2024
Molecular Nano Neural Networks (M3N):In-Body Intelligence for the IoBNT
#InternetofBioNanoThings
#MolecularMachineLearning
#MolecularEngineering
'Bio-Nano Things used in the human body...'
techrxiv.org/users/689760/ar…


1
5
5
413
20 Apr 2024
Molecular Nano Neural Networks (M3N):In-Body Intelligence for the IoBNT
#InternetofBioNanoThings
#IoBNT
#ComputingAndProcessing
#MolecularMachineLearning
#MolecularEngineering
#Nanotechnology
'Bio-Nano Things (BNTs) used in the human body...'
techrxiv.org/users/689760/ar…

1
8
10
2,935