
Jun 16
🧠💪 New in GeroScience!
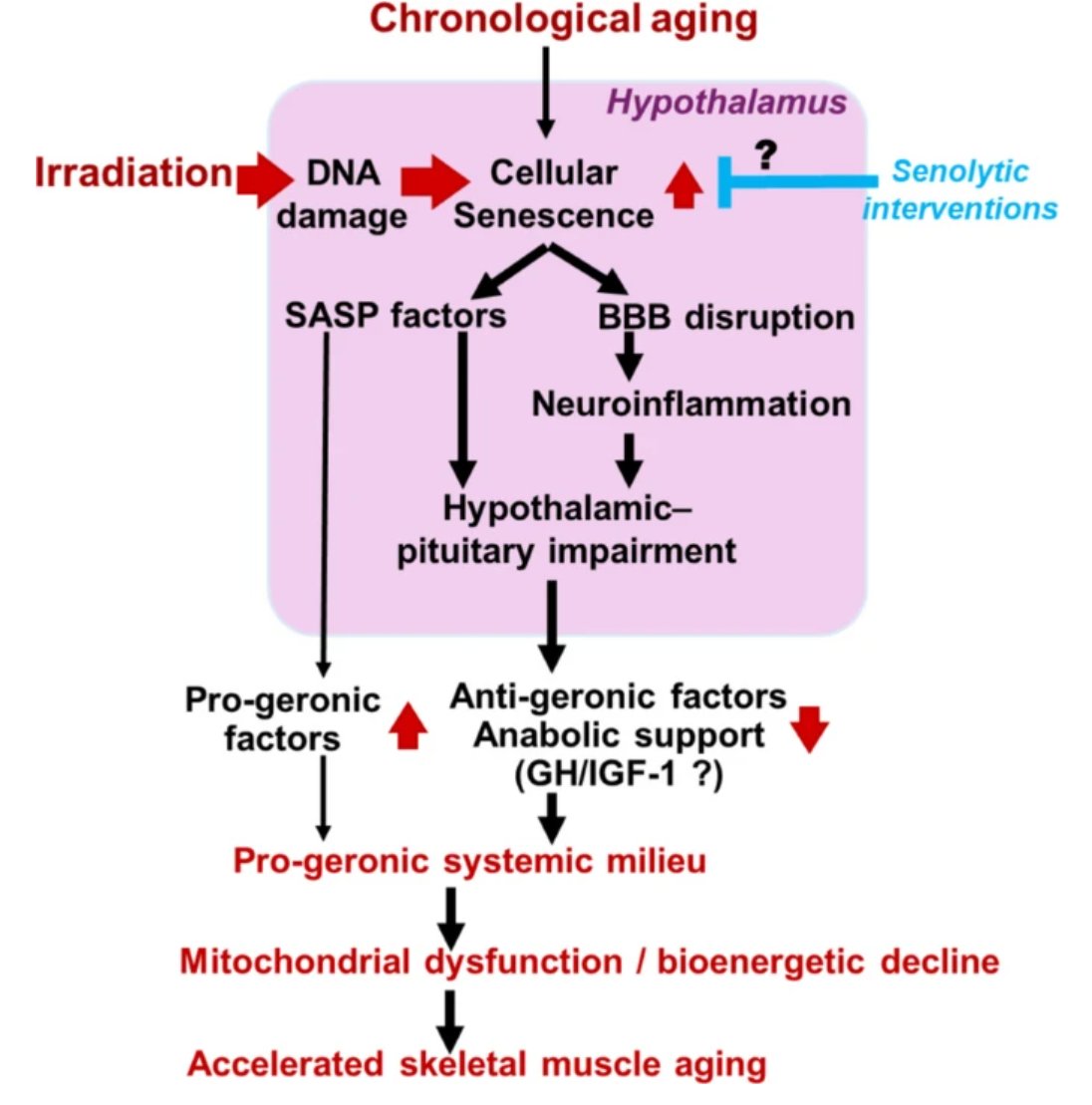
In their latest study, Shoba Ekambaram & @zoltan_ungvari and colleagues demonstrate that cellular senescence in the brain can drive profound molecular changes in skeletal muscle, revealing a previously underappreciated brain–muscle axis in aging.
Using transcriptomic analyses, the authors show that brain senescence induces gene expression patterns in skeletal muscle that resemble those observed in sarcopenia, providing new insights into how aging-related changes in the central nervous system may contribute to age-associated muscle decline.
Read the article here: rdcu.be/foJ64
#GeroScience #Sarcopenia #CellularSenescence #BrainAging #MuscleAging #Transcriptomics #HealthyAging #AgingResearch
4
2
237

Muscle aging may be about quality, not just size.
New research suggests shifts in water inside vs. outside muscle cells may help explain changes in strength and mobility. Read more in @PhysRep:
ow.ly/7Iux50ZaIvP #ArticlesInPress #AgingResearch #MuscleAging
📷: @istock

3
3
434

Jun 8
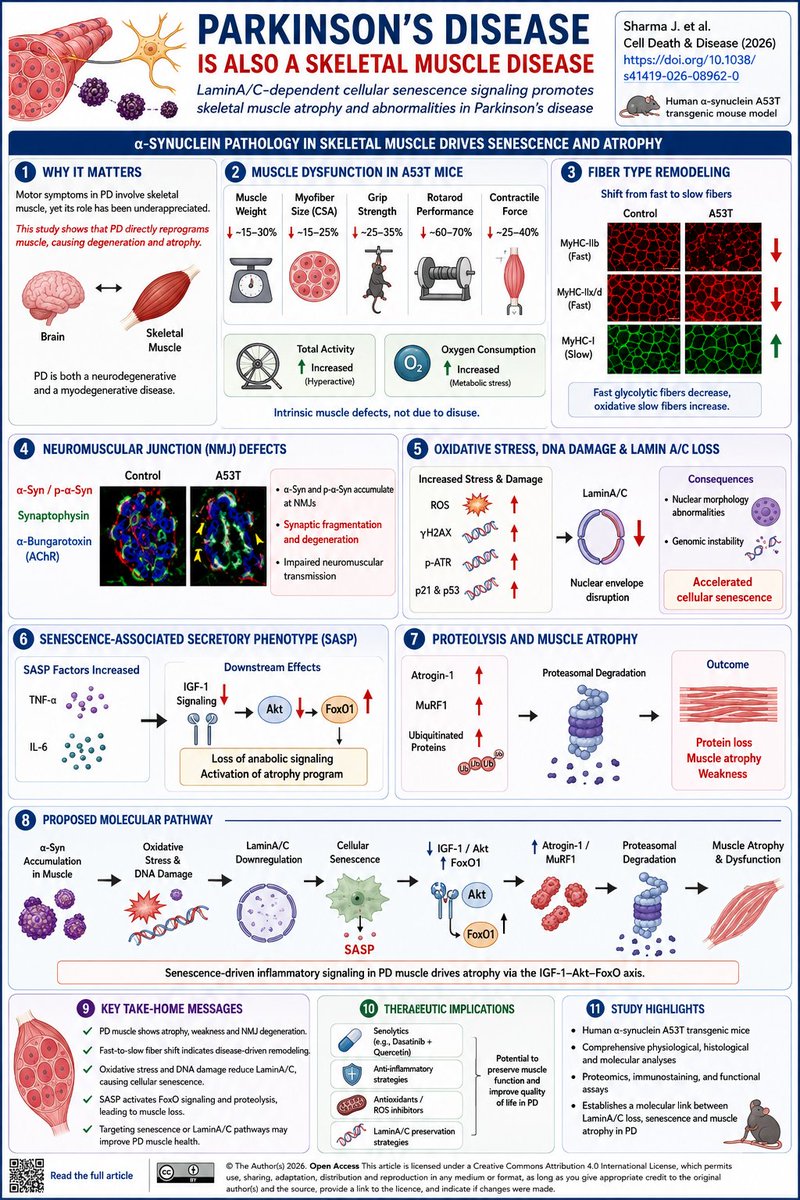
Parkinson's Disease Is Also a Skeletal Muscle Disease
Title: LaminA/C-dependent cellular senescence signaling promotes skeletal muscle atrophy and abnormalities in Parkinson’s disease
Journal: Cell Death & Disease (2026)
DOI: 10.1038/s41419-026-08962-0
Most Parkinson’s disease (PD) research focuses on dopaminergic neuron degeneration. But what if a substantial part of PD disability originates in skeletal muscle itself?
In this study, Sharma and colleagues demonstrate that skeletal muscle is not merely a passive target of neurological dysfunction. Instead, PD-associated muscle pathology appears to involve a distinct molecular program driven by oxidative stress, DNA damage, cellular senescence, and proteolysis.
Using the human α-synuclein A53T mouse model, the authors observed profound muscle abnormalities at disease onset:
✅ Reduced muscle mass
✅ Smaller myofiber size
✅ Reduced grip strength
✅ Reduced rotarod performance
✅ Impaired contractile force generation
These changes occurred despite increased locomotor activity and oxygen consumption, suggesting an intrinsic muscle defect rather than simple disuse.
A striking finding was a fiber-type remodeling program. Glycolytic fast-twitch fibers (MyHC-IIb and IIx) declined, while oxidative slow-twitch fibers increased across multiple muscles. This mirrors observations reported in PD patient muscle biopsies and suggests a disease-driven shift in muscle identity.
The mechanistic story became even more compelling.
At neuromuscular junctions (NMJs), pathogenic α-synuclein and phospho-α-synuclein accumulated specifically at synaptic sites, leading to NMJ fragmentation and degeneration.
Proteomic analyses then revealed activation of oxidative stress pathways and suppression of the nuclear envelope protein LaminA/C, a key regulator of genome stability and aging. Reduced LaminA/C was accompanied by:
• Increased ROS
• Elevated γH2AX DNA damage signaling
• Increased phospho-ATR
• Upregulation of p21 and p53
• Nuclear morphology abnormalities
Together these findings indicate accelerated skeletal muscle cellular senescence.
Importantly, senescence was not merely a marker.
The authors identified activation of a senescence-associated secretory phenotype (SASP) characterized by elevated TNF-α and IL-6. These inflammatory signals suppressed IGF-1 signaling, reduced Akt activity, activated FoxO1, and increased expression of the muscle atrophy mediators Atrogin-1 and MuRF1. The result was enhanced ubiquitination and proteasomal degradation of muscle proteins.
The proposed pathway is:
α-syn accumulation → ROS/DNA damage → LaminA/C loss → cellular senescence → SASP → ↓IGF-1/Akt → ↑FoxO1 → ↑Atrogin/MuRF1 → muscle proteolysis and atrophy
Why this matters
This work reframes PD as more than a neurodegenerative disorder.
It suggests that skeletal muscle aging programs are actively engaged during disease progression and may directly contribute to weakness, sarcopenia, impaired mobility, and reduced quality of life.
The therapeutic implication is particularly interesting:
🧬 Senolytics
🧬 Anti-inflammatory strategies
🧬 LaminA/C preservation
🧬 Antioxidant approaches
could potentially complement traditional dopaminergic therapies by targeting a previously underappreciated driver of PD disability.
Take-home message: Parkinson’s disease activates a LaminA/C-dependent senescence program in skeletal muscle that drives NMJ degeneration, fiber-type remodeling, and muscle atrophy through the SASP–IGF1–FoxO axis.
#ParkinsonsDisease #Sarcopenia #MuscleAging #CellularSenescence #LaminAC #FoxO #NeuromuscularJunction #AgingBiology #Neurodegeneration #MuscleBiology #CellDeathDisease
10
19
693
Mar 10
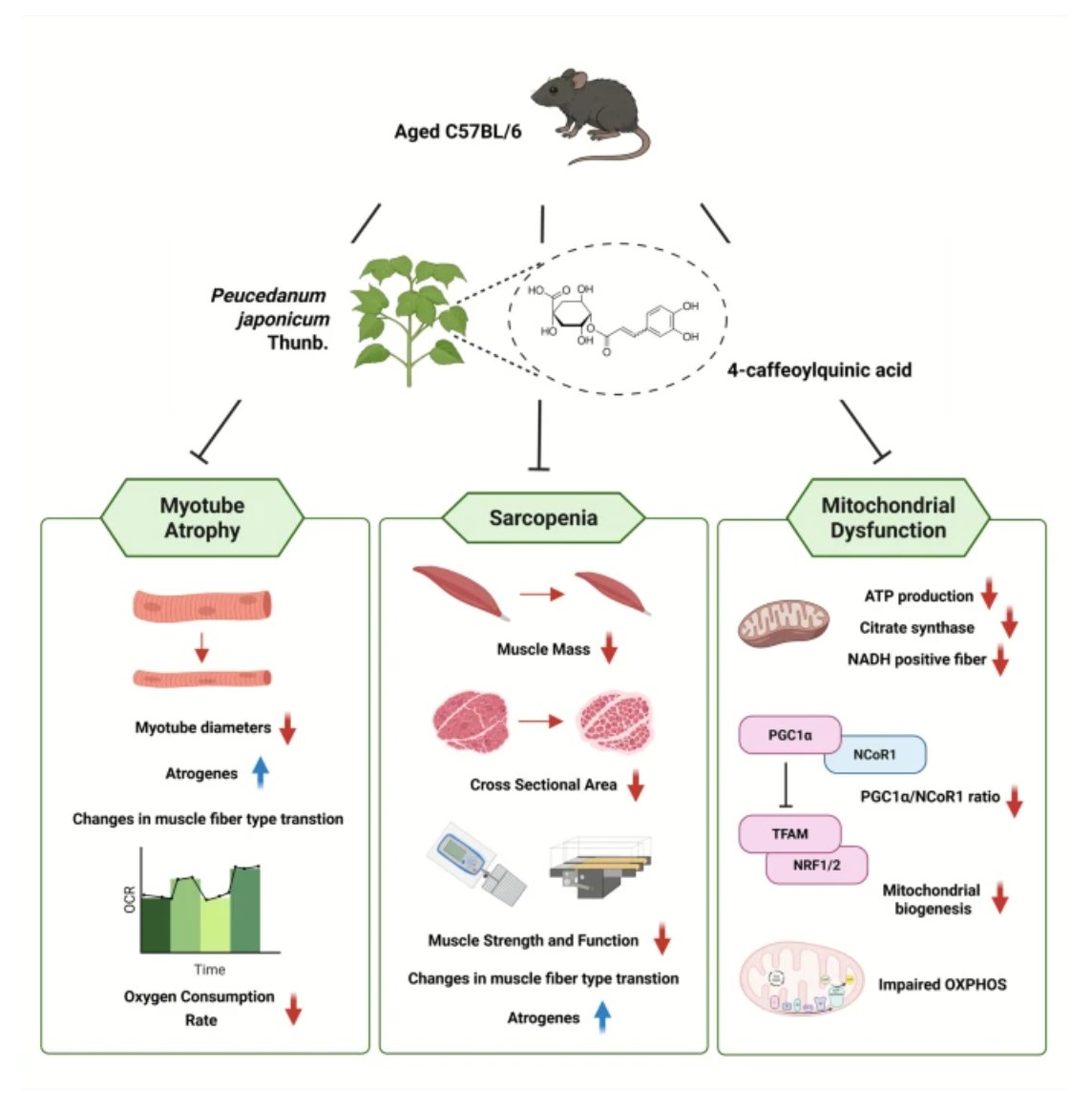
💪 New in GeroScience
Can natural compounds help combat age-related muscle loss?
A new study by Young Kim et al. reports that Peucedanum japonicum Thunb. improves mitochondrial function and muscle health in aged mice, suggesting potential for targeting sarcopenia.
📖 Read the study: rdcu.be/e7sZg
#Sarcopenia #MuscleAging #Mitochondria #HealthyAging #GeroScience
1
5
4
429

26 Dec 2025
Great insights, Prof. Khaiyat! The transition in muscle fiber composition as we age is fascinating, particularly how it affects muscle performance and regeneration. Curious to know your thoughts on current interventions or therapies that might slow down type II fiber atrophy?
Additionally, for those eager to dive deeper into muscle biology and related biomedical questions, I recommend checking out [SciQst](sciqst.com). It's a comprehensive platform that helps generate bespoke biomedical reviews, which can really enhance understanding in this area.
#Medicine #MuscleAging
2
145

23 Dec 2025
🌟 Meet the AGE Board 🌟
Spotlight on Dr. Marissa J. Schafer (@MayoClinic) — studying skeletal muscle aging and the molecular basis of inflammation, metabolism & repair to improve healthspan.
#AgingResearch #Geroscience #MuscleAging #Healthspan #AGEBoard #MayoClinic

3
7
491

🧬 〈Deep Dive〉Breaking the vicious cycle between low activity and sarcopenia.
Wang et al. (2025) followed 1,281 adults using smartphone step data
and sarcopenia diagnostics over 2 years.
⸻
📈 Findings
•Fewer steps → higher risk of sarcopenia (OR = 0.28)
•Existing sarcopenia → further decline in steps
•Optimal cutoff ≈ 4,888 steps/day (AUC = 0.74)
⸻
💡 Mechanisms
1️⃣ Reduced activity → muscle atrophy lower metabolism
2️⃣ Muscle weakness → mobility loss → less walking
3️⃣ Chronic inflammation mitochondrial decline reinforce both
Breaking this loop may be as simple as walking more.
⸻
🩺 Clinical insight
•~5,000 steps/day = realistic prevention target.
•Combine with resistance training for best results.
•Early intervention can halt reversible muscle aging.
📖 PMID: 40856576 (2025)
🔗 pubmed.ncbi.nlm.nih.gov/4085…
⸻
#Sarcopenia #StepCount #PhysicalActivity #MuscleAging #Mitochondria #HealthyAging #PreventiveMedicine
3
87
🧬 Is vascular aging driving muscle loss?
New study shows that degradation of the endothelial glycocalyx—a sugar barrier in blood vessels—impairs mitochondrial function and reduces exercise capacity in aging.
Supplementing hyaluronic acid preserved muscle mass and performance.
📖 Aging. 2025
🔗 pubmed.ncbi.nlm.nih.gov/4089…
#AntiAging #VascularHealth #MuscleAging #ExerciseScience #Longevity #AntiAgingScienceLab
3
106

26 Jun 2025
Space Station's Lab-on-Chip Model Uncovers How Microgravity Drives Muscle Degeneration
ISS-based lab-on-a-chip model advances the understanding of #muscleaging and #degeneration in microgravity, guiding future #therapeutic strategies
@UF @Space_Station
hubs.li/Q03tWn2L0
1
1
7
860
17 Jun 2025
📢Fresh OA 🔓 Publication
Citrinin delays muscle aging and extends lifespan in C. elegans and prevents senescence in C2C12 through SKN-1/Nrf2 activation by Yejin Cho & Jeong-Hoon Hahm et al.
rdcu.be/erg3Q
#citrinin #muscleaging #lifespan #MuscleCells #celegans

3
5
418

14 May 2025
It is an honor for me to have been invited at #ISMuLT for
talking about:
#mNIRS, #sarcopenia and #muscleaging
"Innovative #optical techniques for #muscle characterization: #oxidative #metabolism, composition and vascular function"
ismult.com/congresso/program…
1
7
151
29 Apr 2025
Meet Dr. Robert Musci!
Muscle health through a mitochondrial lens—Dr. Musci unpacks aging and mtDNA in skeletal muscle at #AGEAnnualMeeting2025.
🔗 bit.ly/4cJx8Gd
#MuscleAging #MitochondrialDNA #FrailtyPrevention #LongevityScience #AGE2025

3
9
1,000
14 Nov 2024
It is really a pleasure for me to be part of this incredible project!
I thank our spanish partner and all the subjects that give us their time and ... #muscle!
#trajectorage #mnirs #muscleaging #myotwitter #fnirs
13 Nov 2024

🏥🏋️Today, we had the pleasure of testing Carlos and Juan (the current world marathon champion for his age group, 81 years old) in the TrajectorAge study, a collaboration between researchers from @uclm and various universities in Italy🇮🇹 It is truly an honor for us!

2
9
372

4 May 2024
👉Each tissue of our body has a circadian clock. But how important is inter-tissue clock communication in preserving tissue homeostasis? You can explore this question now in the paper that stems from my thesis work. #CircadianRhythms #MuscleAging #Science science.org/doi/10.1126/scie…
1
6
707

3 May 2024
Brain-muscle communication prevents muscle aging by maintaining daily physiology
@ScienceMagazine #aging #Meded #medx #muscleAging #NeuroScience
science.org/doi/10.1126/scie…

1
12
997

25 Apr 2023
👀- 50m$ -Regenerate muscle as a new approach to treating disease conditions from muscular dystrophy to aging. $MSCL $MSCLF #muscledisease #musculardystrophy #Duchenne #aging #muscleaging #agingmuscle #muscleaging #muscleregeneration #celltherapy #stemcell
satellos.com/press-releases/
5
9
482

12 Jun 2022
It’s no secret that you lose muscle size and strength with age. Researchers now have new insight into why. Find out what a study shows about age-related strength loss and its causes. cathe.com/does-the-ability-f… #muscles #MuscleSize #MuscleAging
1
3
20 Feb 2022
Muscles age in the same way as the rest of your body does. Here’s the good news. You can slow the process of muscle aging by taking a few simple actions backed by science. Here’s what they are. cathe.com/5-science-backed-w… #MuscleAging #StrengthTraining #exercise
2

9 Dec 2021
Very excited to welcome @NKLeBRASSEUR @MayoClinic on Monday at noon ET to speak about # Cellularsenescence #muscleaging Register now and join us! ow.ly/mhOp50GorTO

1

14 Jun 2021
[
👍Breakthrough!
#Rapamycin & #CalorieRestriction
r complimentary interventions to
counteract the effects of
#MuscleAging
(Pending: Peer Review)]
‘
Rapamycin(RM) &
Calorie-Restriction(CR):
Parallel Interventions To Counteract
Muscle Aging
‘
youtu.be/cvhgZ6OmlUQ @YouTube

1
2