
Day 10 of the #MDSimulation workshop: protein-ligand complex pipeline completed in #GROMACS. Topology merged, restraints applied, production MD run confirmed.Participants independently ran kinase-imatinib simulation and presented full #MDAnalysis outputs.
bdglifesciences.com/event/md…

1
Day 9 of the MD Simulation workshop: five analysis parameters completed — RMSD, RMSF, hydrogen bonds, radius of gyration, potential energy. Then: protein-ligand complex preparation begins with 3HTB.
One day left.
#GROMACS #MDSimulation #ComputationalBiology

9
Day 8 of the #MDSimulation workshop: full #GROMACS pipeline completed. Ionisation, energy minimisation, NVT, NPT, production MD run — six output files confirmed. Current publication standard: 400–500 ns. Participants now know exactly what that means & why.
#ComputationalBiology

6
Day 7 of the #MDSimulation workshop: #GROMACS phase begins. Lysozyme topology generated with OPLS-AA, water box solvated with 12,596 TIP3P molecules, topology file verified at every step.
This is where most MD courses start. This workshop is already on Day 7.

7
Day 6 of the MD Simulation workshop: Linux environment configured, GROMACS installed via WSL, gmx verified in the Ubuntu terminal. The Windows phase is complete.
Protein-ligand complex simulations in GROMACS begin in two days.
#GROMACS #MDSimulation #ComputationalBiology

24
Day 4 of the #MDSimulation workshop: participants ran their first NAMD simulations. Sphere done. Box done. 11 output files generated per run. The log file is ready for analysis.
Troubleshooting path errors and missing DLL files was part of the curriculum too.
#DrugDiscovery

6
Day 3 of the #MDSimulation workshop: water box solvation completed, both boundary conditions visualised in VMD, and the NAMD configuration file walked through line by line.
Every input file is in place. Day 4 is the simulation run.
#ComputationalBiology #DrugDiscovery

11

Run MD simulation on your iPad or Cloud, no install needed! 🚀
Fully browser-based
Auto-analysis: RMSD, Free Energy, & publication-ready plots~
For education and research!
#compchem #MDsimulation #chemcom #bioinfomatics
@JCIM_JCTC asap!
pubs.acs.org/doi/10.1021/acs…
2
29
138
6,732
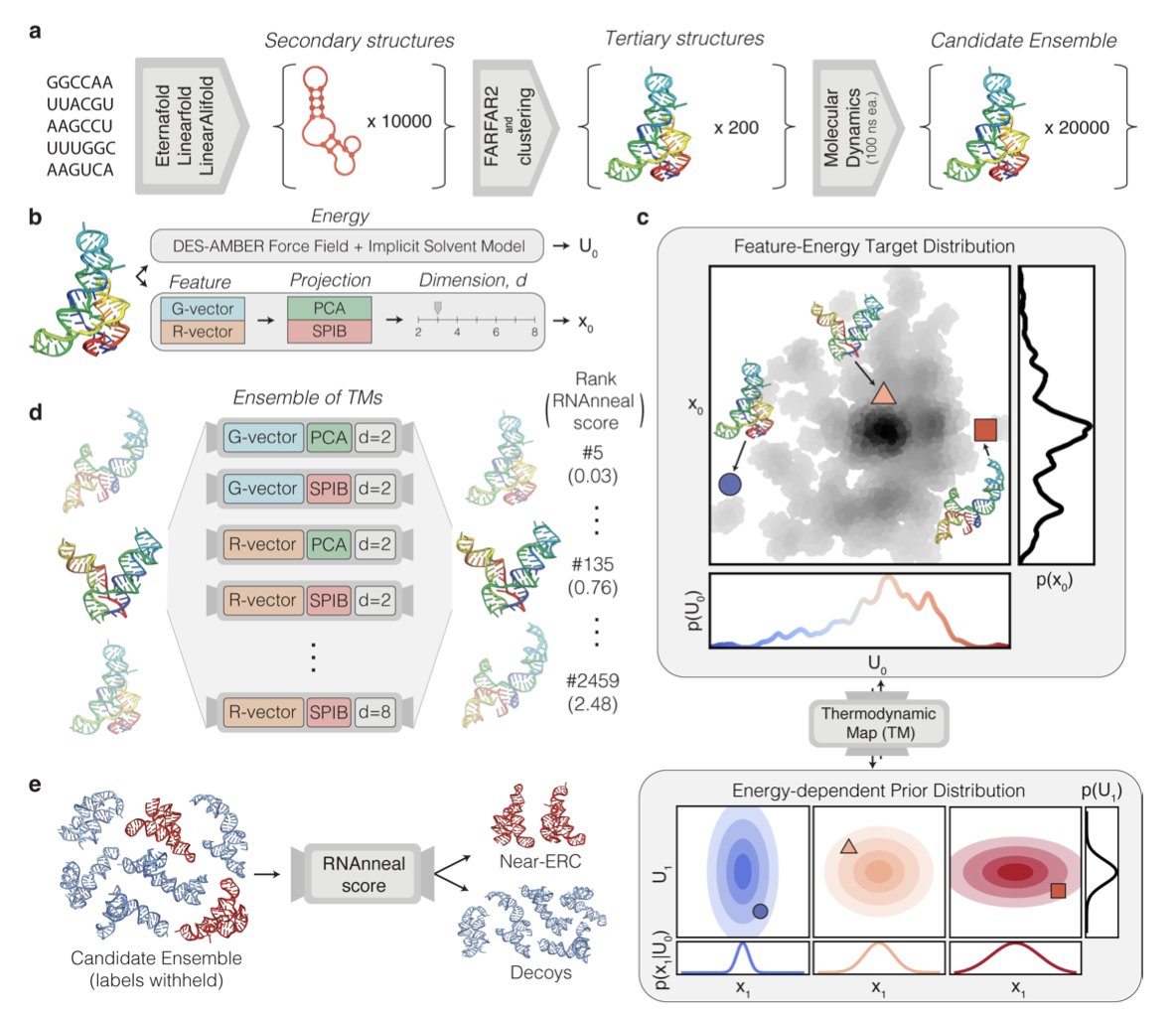
Ab initio prediction of RNA structure ensembles with RNAnneal
1 RNAnneal predicts full 3D RNA ensembles from sequence alone—no experimental restraints, no homology templates, no supervised training data.
2 It fuses generative deep learning (Thermodynamic Maps) with physics: first‐principles secondary‐structure sampling, FARFAR2 3D assembly, 100 ns implicit-solvent MD, then unsupervised scoring.
3 An ensemble of 28 diffusion‐based Thermodynamic Maps learns the joint distribution of latent geometries MD energies; the consensus “RNAnneal score” ranks every snapshot by native-likeness.
4 Benchmarked on 16 riboswitch crystal structures: median AUAC 0.83 vs 0.67 (DES-AMBER) & 0.59 (Rosetta); BEDROC 0.40 vs 0.26 & 0.16—without ever seeing an RNA structure during training.
5 Pseudoknot-free riboswitches are recovered with <10 Å RMSD or >0.75 INF in 11/16 cases; pseudoknot systems remain challenging but are still discriminated from decoys.
6 Introduces per-nucleotide “interaction entropy” that quantifies how many distinct base-pair/stacking states each residue visits—turning ensemble heterogeneity into a colored 1D track.
7 Temperature annealing of the generative prior further boosts ERC detection for 11/16 RNAs, showing direct control over energy‐landscape exploration.
8 Holo and apo riboswitch ensembles are automatically split into weighted conformational states; top representatives often match the ligand-bound crystal within 5 Å RMSD.
9 Entropy hotspots coincide with ligand/metal binding sites, hinting at allosteric switches—useful for RNA drug-discovery campaigns that need dynamic pockets, not single frozen poses.
10 Fully modular pipeline: any new secondary‐structure sampler, MD engine, or diffusion architecture can be plugged in; Mg²⁺ ions & pseudoknot templates are next targets.
💻Code: go.umd.edu/rnanneal
📜Paper: biorxiv.org/content/10.64898…
#RNA #RNAstructure #riboswitch #diffusionmodels #abinitio #MDsimulation #drugdiscovery
2
7
52
3,117
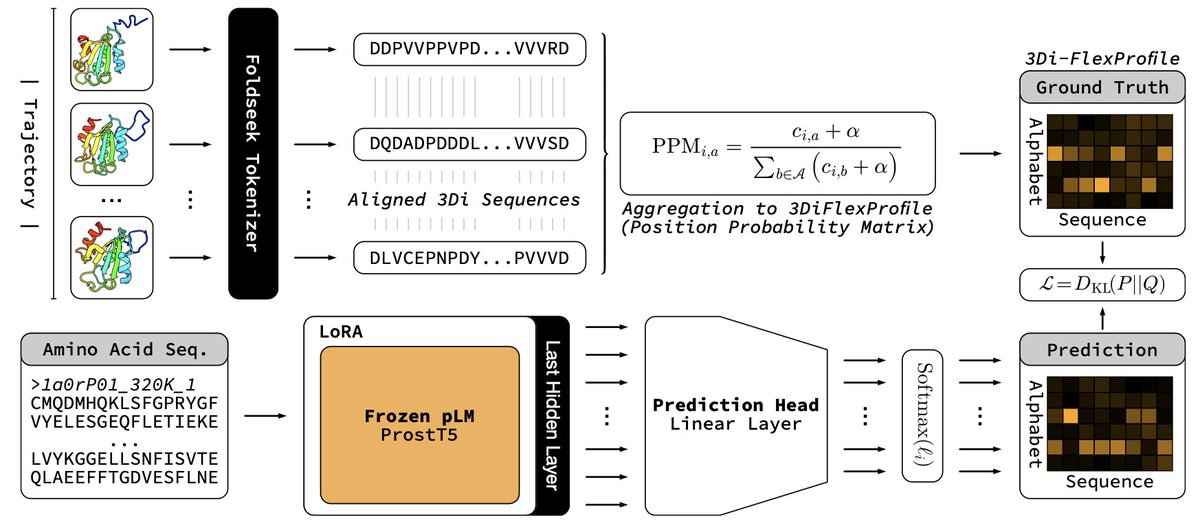
Protein Language Modeling Beyond Static Folds Reveals Sequence-Encoded Flexibility
1 ProtProfileMD translates a single amino-acid sequence into a per-residue probability profile over 3Di structural tokens, capturing how often each local conformation is visited during MD without running any simulation at query time.
2 The model distills 5 398 CATH domains’ 320 K trajectories into a 3Di “FlexProfile”; entropy of the predicted profile correlates with RMSF at PCC 0.60, flagging flexible or disordered segments genome-wide in seconds.
3 Supervised LoRA fine-tuning of ProstT5 (only 2.2 M trainable params) lets the pLM learn an implicit energy-weighted ensemble, bypassing costly all-atom ensemble generation while staying proteome-scalable (~0.1 s/protein).
4 Remote homology detection benchmarked on SCOPe shows ROC-AUC gains at Family, Superfamily and Fold levels over standard 3Di search, proving that dynamics fingerprints boost sensitivity beyond static structure tokens.
5 Framework is alphabet-agnostic: any structural vocabulary (PB, DSSP, internal coords) or experimental ensemble (cryo-EM, NMR) can replace 3Di, making FlexProfiles a generic route to encode motion inside language models.
💻Code: github.com/finnlueth/ProtPro…
📜Paper: biorxiv.org/content/10.64898…
#proteindynamics #proteinlanguageModel #computationalBiology #bioinformatics #mdSimulation #structuralAlphabet #homologyDetection
6
31
2,023

24 Sep 2025
【技術書更新!】
📘 第5章:リガンド-タンパク質 MDsimulation(分子動力学シミュレーション)
zenn.dev/labcode/books/1e05d…
にて、GROMACSの環境構築の仕方をアップデートしました!めちゃくちゃ楽になったので、ぜひご確認ください!
1
4
525

27 Aug 2025
📢Congrats to Jiangtao Guo on their study "Structural basis for the ion selectivity of potassium-chloride cotransporter KCC4 revealed by cryo-EM titration" published in Biophysics Reports (Online First).
🔗doi.org/10.52601/bpr.2025.24…
#KCC4 #CryoEMTitration #MDsimulation

ALT Figure 1. High-resolution structures of KCC4 in different ion conditions. A 3D reconstruction of KCC4KNO3 colored by local resolution. B Sample of EM maps at transmembrane helices 1 (TM1) and 6 (TM6) of KCC4 in different ion conditions at the contour level of 4 σ. C Cartoon representation of one KCC4KCl subunit (left) and the zoom-in view of three potential ion binding sites S1–S3 (right). S1–S3 are equivalent to the three sites (SK, SCl1, SCl2) that we initially assigned in the KCC16KKR structure. The ions are shown as cyan spheres
1
2
130

23 Jul 2025
⚡GeNNIP4MDを用いたニューラルネットワーク力場で、プロピレングリコールの誘電特性を高精度に再現!低周波帯の解析も可能にし、材料設計を革新します。
🔍 詳細はTECHBLOGをご覧ください ▶ blog.fltech.dev/entry/2025/0… #GeNNIP4MD #MDsimulation #誘電関数 #次世代材料設計
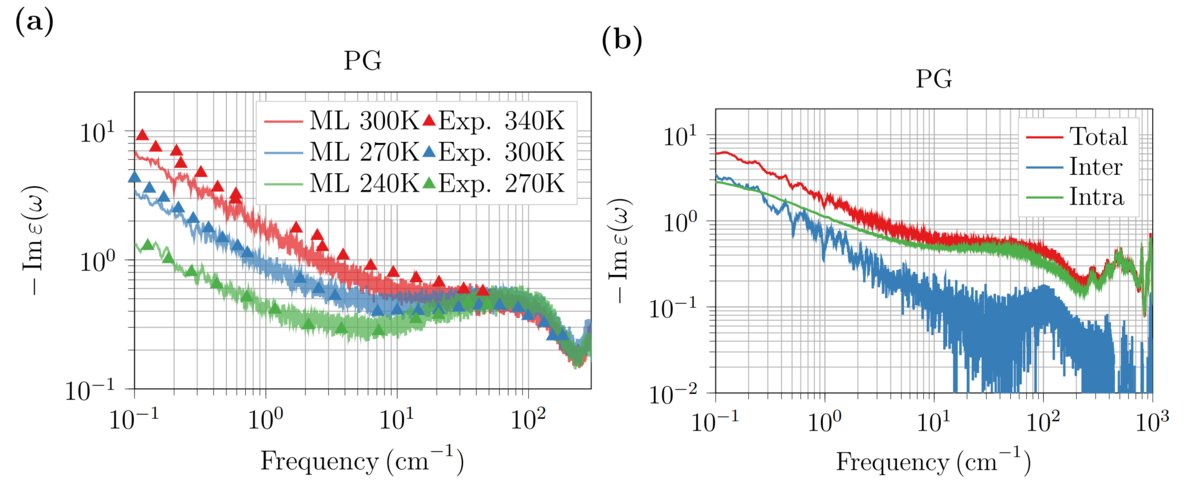
ALT (a) 3つの異なる温度(240 K(緑)、270 K(青)、300 K(赤))のPGの誘電関数。(b) 300Kの誘電関数を系全体(赤)と分子内の成分(緑)、および分子間成分(青)に分けた結果。
4
149
8 Jun 2025
【技術書一部公開!】
第5章:リガンド-タンパク質 MDsimulation(分子動力学シミュレーション)
MD simulationについて、一からほぼコピペでできるように書いています!ここからぜひMD simulationを始めてみてください!
この内容が載っている技術書はこちら!
zenn.dev/labcode/books/1e05d…
4
17
814

27 May 2025
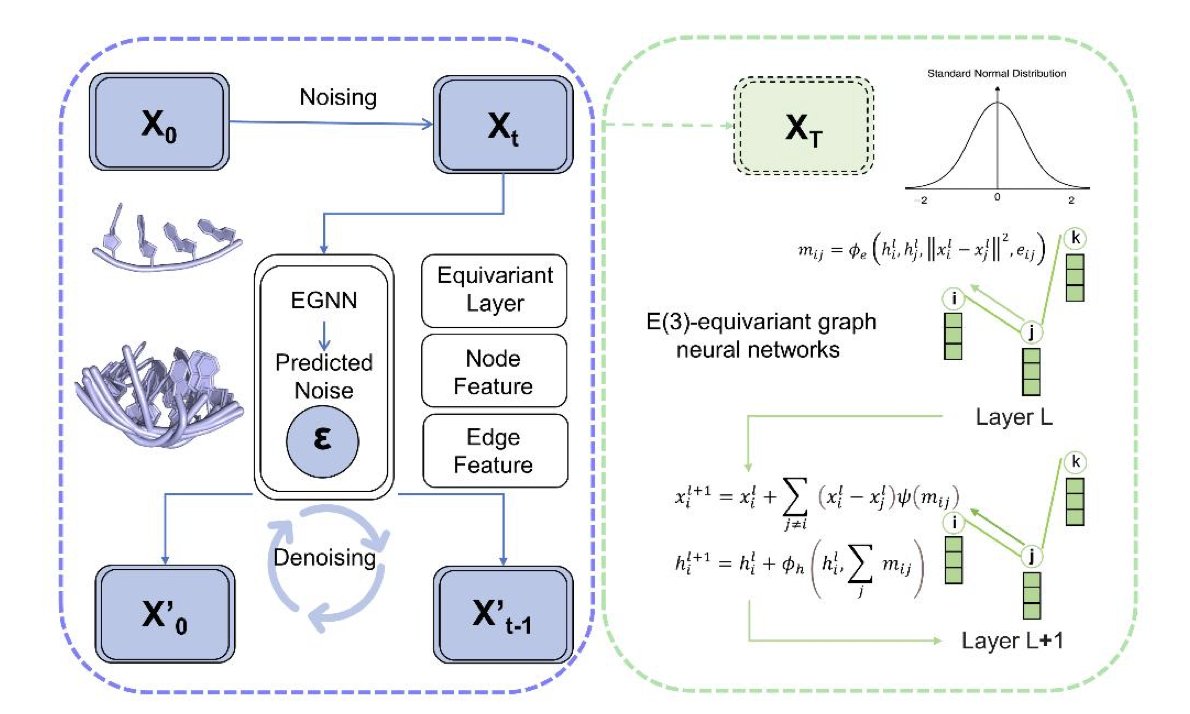
DynaRNA: Dynamic RNA Conformation Ensemble Generation with Diffusion Model
1.This paper introduces DynaRNA, the first diffusion-based generative model to simulate RNA conformation ensembles directly in 3D coordinate space, achieving high geometric fidelity and capturing RNA's structural dynamics orders of magnitude faster than molecular dynamics (MD) simulations.
2.DynaRNA leverages a denoising diffusion probabilistic model (DDPM) with Equivariant Graph Neural Networks (EGNNs), enabling end-to-end generation of RNA conformations while preserving E(3) symmetry and avoiding the need for multiple sequence alignment (MSA).
3.Using partial noising instead of full corruption allows DynaRNA to balance diversity and structural fidelity, making it well-suited for modeling heterogeneous and flexible RNA conformational landscapes.
4.In benchmarking on tetranucleotides, DynaRNA outperforms traditional MD simulations by generating ensembles with intercalation rates below 10%, compared to >90% in OL3 force field simulations, more closely matching NMR experimental results.
5.DynaRNA captures dihedral angle distributions and local structural metrics such as C4'-C4' distances and hyper bond angles with high accuracy, validating its physical plausibility relative to PDB structures.
6.The model successfully captures rare and transient RNA excited states, including the low-population ES2 of HIV-1 TAR (~0.4% occupancy), demonstrating its ability to overcome energy barriers that challenge traditional MD simulations.
7.DynaRNA can de novo fold RNA tetraloops from extended single-stranded RNA sequences without structural restraints, achieving atomic RMSD as low as 0.9 Å compared to experimentally determined native structures.
8.Compared to MD, DynaRNA dramatically reduces computational cost, producing high-quality conformational ensembles in minutes on a single GPU versus weeks of simulation time.
9.Its flexible, data-driven nature makes DynaRNA amenable to hybrid frameworks, such as combining AI-generated ensembles with short MD refinement or integrating richer atomic representations and RNA force fields.
10.This framework opens new avenues for modeling RNA conformational heterogeneity, RNA-targeted drug discovery, RNA-protein interaction prediction, and the design of RNA-based therapeutics including mRNA vaccines.
11.DynaRNA marks a paradigm shift by transitioning from static structure prediction to dynamic ensemble modeling in RNA, extending the generative modeling revolution initiated by AlphaFold to the realm of RNA dynamics.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #DiffusionModel #GenerativeBiology #StructuralBioinformatics #RNAfolding #EGNN #MDsimulation #RNAtherapeutics #ConformationalDynamics #ComputationalBiology

9
773
27 May 2025
DynaRNA: Dynamic RNA Conformation Ensemble Generation with Diffusion Model
1.This paper introduces DynaRNA, the first diffusion-based generative model to simulate RNA conformation ensembles directly in 3D coordinate space, achieving high geometric fidelity and capturing RNA's structural dynamics orders of magnitude faster than molecular dynamics (MD) simulations.
2.DynaRNA leverages a denoising diffusion probabilistic model (DDPM) with Equivariant Graph Neural Networks (EGNNs), enabling end-to-end generation of RNA conformations while preserving E(3) symmetry and avoiding the need for multiple sequence alignment (MSA).
3.Using partial noising instead of full corruption allows DynaRNA to balance diversity and structural fidelity, making it well-suited for modeling heterogeneous and flexible RNA conformational landscapes.
4.In benchmarking on tetranucleotides, DynaRNA outperforms traditional MD simulations by generating ensembles with intercalation rates below 10%, compared to >90% in OL3 force field simulations, more closely matching NMR experimental results.
5.DynaRNA captures dihedral angle distributions and local structural metrics such as C4'-C4' distances and hyper bond angles with high accuracy, validating its physical plausibility relative to PDB structures.
6.The model successfully captures rare and transient RNA excited states, including the low-population ES2 of HIV-1 TAR (~0.4% occupancy), demonstrating its ability to overcome energy barriers that challenge traditional MD simulations.
7.DynaRNA can de novo fold RNA tetraloops from extended single-stranded RNA sequences without structural restraints, achieving atomic RMSD as low as 0.9 Å compared to experimentally determined native structures.
8.Compared to MD, DynaRNA dramatically reduces computational cost, producing high-quality conformational ensembles in minutes on a single GPU versus weeks of simulation time.
9.Its flexible, data-driven nature makes DynaRNA amenable to hybrid frameworks, such as combining AI-generated ensembles with short MD refinement or integrating richer atomic representations and RNA force fields.
10.This framework opens new avenues for modeling RNA conformational heterogeneity, RNA-targeted drug discovery, RNA-protein interaction prediction, and the design of RNA-based therapeutics including mRNA vaccines.
11.DynaRNA marks a paradigm shift by transitioning from static structure prediction to dynamic ensemble modeling in RNA, extending the generative modeling revolution initiated by AlphaFold to the realm of RNA dynamics.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #DiffusionModel #GenerativeBiology #StructuralBioinformatics #RNAfolding #EGNN #MDsimulation #RNAtherapeutics #ConformationalDynamics #ComputationalBiology
3
14
1,097
22 May 2025
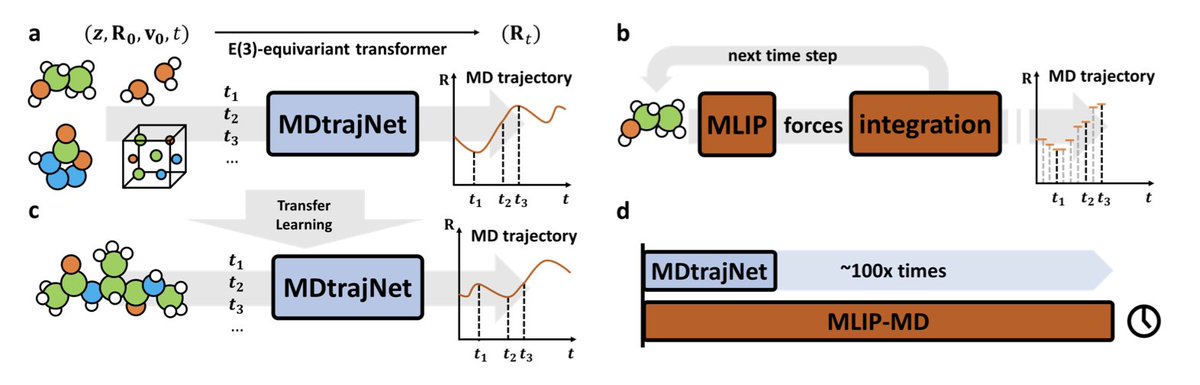
Artificial Intelligence for Direct Prediction of Molecular Dynamics Across Chemical Space
1.MDtrajNet-1 introduces a paradigm shift in molecular dynamics (MD) by directly predicting atomic trajectories in 4D spacetime—bypassing force calculations and time-step integration. This foundational model enables simulations that are over 100× faster than traditional MD, even those accelerated by ML force fields.
2.The model replaces iterative propagation with a Transformer-based, E(3)-equivariant neural network that learns to map atomic positions, velocities, element types, and time intervals directly to future geometries. This allows large-scale parallelization and efficiency without sacrificing accuracy.
3.MDtrajNet-1 achieves remarkable accuracy: in short-term predictions (10 fs), it reaches sub-picometer error levels, rivaling ab initio MD. For long-term predictions (10 ps), it reproduces spectral features with fidelity comparable to DFT-level simulations, despite being trained on a fraction of the data.
4.The model is trained on 1 million data points sampled from the ANI-1xMD dataset, covering 173 molecular systems with up to 9 atoms. Despite the limited size and diversity, MDtrajNet-1 generalizes well across unseen molecules and chemical spaces.
5.Compared to the GICnet model (an earlier proof-of-concept), MDtrajNet-1 demonstrates superior accuracy, scalability, and generalizability, thanks to architectural advances like multi-head equivariant attention and fine-grained representation of local atomic environments.
6.The model supports flexible ensemble conditions. Beyond NVE, MDtrajNet-1 can be retrained for NVT simulations and yields high-quality vibrational spectra even with added complexity from thermostats.
7.Its performance extends to periodic systems and different interaction types. It successfully simulates diamond lattices and Lennard-Jones fluids under periodic boundary conditions, capturing structural features like radial distribution functions.
8.MDtrajNet-1 also exhibits strong transfer learning capabilities. When fine-tuned on short trajectories from a 22-atom alanine dipeptide, it reproduces long-timescale conformational dynamics (Ramachandran plots) more accurately than MLIPs trained on the same data.
9.The model architecture is O(3)-equivariant, atom-centered, and scalable with system size. Computational time grows linearly with atom number, enabling efficient simulations of large systems on standard hardware (e.g., RTX 4090).
10.MDtrajNet-1 paves the way for general-purpose, foundational models in molecular simulation. Its blend of physics-inspired architecture and generative capability offers a compelling path toward scalable, accurate, and efficient trajectory generation in chemical and materials science.
💻Code: github.com/dralgroup/mlatom
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularDynamics #4DSpacetime #TransformerModel #EquivariantNN #MDsimulation #AI4Science #ComputationalChemistry #MDtrajNet

3
17
1,150
22 May 2025
Artificial Intelligence for Direct Prediction of Molecular Dynamics Across Chemical Space
1.MDtrajNet-1 introduces a paradigm shift in molecular dynamics (MD) by directly predicting atomic trajectories in 4D spacetime—bypassing force calculations and time-step integration. This foundational model enables simulations that are over 100× faster than traditional MD, even those accelerated by ML force fields.
2.The model replaces iterative propagation with a Transformer-based, E(3)-equivariant neural network that learns to map atomic positions, velocities, element types, and time intervals directly to future geometries. This allows large-scale parallelization and efficiency without sacrificing accuracy.
3.MDtrajNet-1 achieves remarkable accuracy: in short-term predictions (10 fs), it reaches sub-picometer error levels, rivaling ab initio MD. For long-term predictions (10 ps), it reproduces spectral features with fidelity comparable to DFT-level simulations, despite being trained on a fraction of the data.
4.The model is trained on 1 million data points sampled from the ANI-1xMD dataset, covering 173 molecular systems with up to 9 atoms. Despite the limited size and diversity, MDtrajNet-1 generalizes well across unseen molecules and chemical spaces.
5.Compared to the GICnet model (an earlier proof-of-concept), MDtrajNet-1 demonstrates superior accuracy, scalability, and generalizability, thanks to architectural advances like multi-head equivariant attention and fine-grained representation of local atomic environments.
6.The model supports flexible ensemble conditions. Beyond NVE, MDtrajNet-1 can be retrained for NVT simulations and yields high-quality vibrational spectra even with added complexity from thermostats.
7.Its performance extends to periodic systems and different interaction types. It successfully simulates diamond lattices and Lennard-Jones fluids under periodic boundary conditions, capturing structural features like radial distribution functions.
8.MDtrajNet-1 also exhibits strong transfer learning capabilities. When fine-tuned on short trajectories from a 22-atom alanine dipeptide, it reproduces long-timescale conformational dynamics (Ramachandran plots) more accurately than MLIPs trained on the same data.
9.The model architecture is O(3)-equivariant, atom-centered, and scalable with system size. Computational time grows linearly with atom number, enabling efficient simulations of large systems on standard hardware (e.g., RTX 4090).
10.MDtrajNet-1 paves the way for general-purpose, foundational models in molecular simulation. Its blend of physics-inspired architecture and generative capability offers a compelling path toward scalable, accurate, and efficient trajectory generation in chemical and materials science.
💻Code: github.com/dralgroup/mlatom
📜Paper: doi.org/10.26434/chemrxiv-20…
#MolecularDynamics #4DSpacetime #TransformerModel #EquivariantNN #MDsimulation #AI4Science #ComputationalChemistry #MDtrajNet
1
12
991
1 Mar 2025
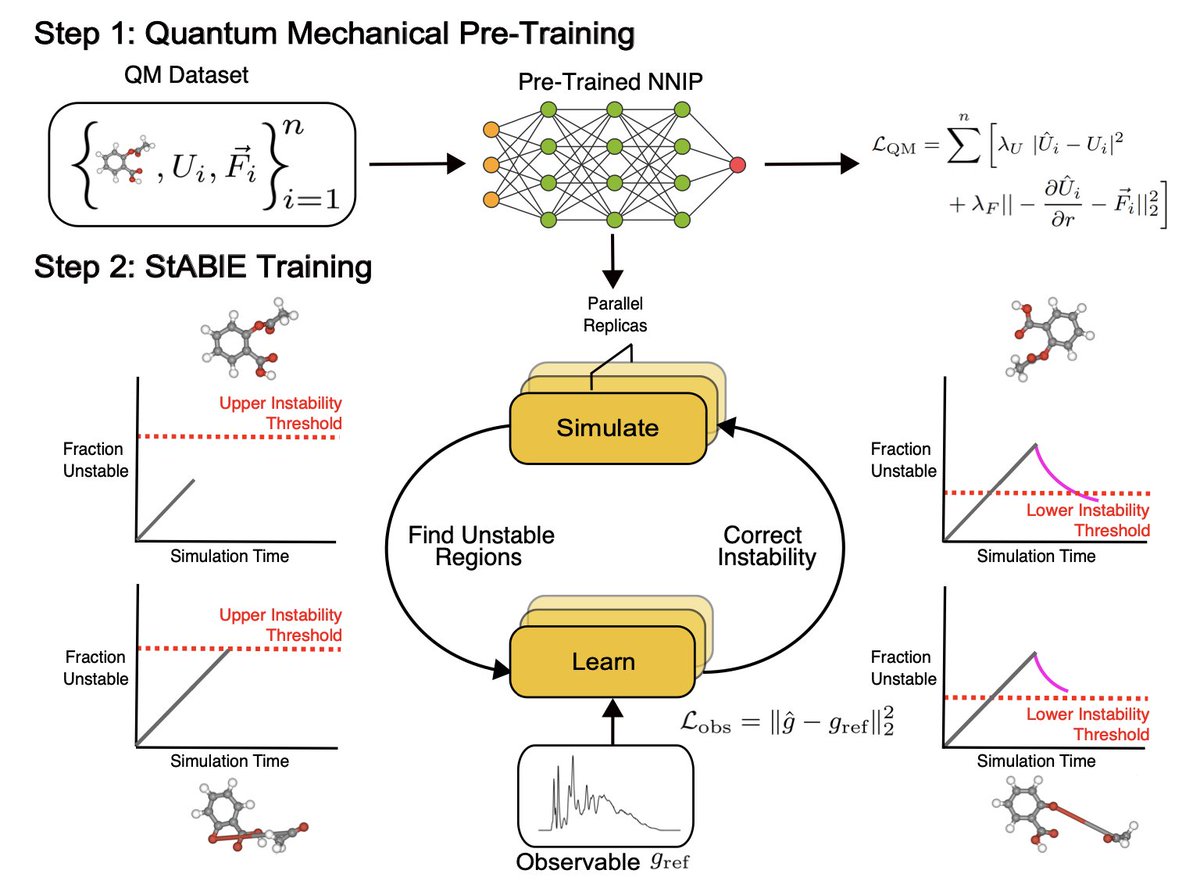
Stability-Aware Training of Machine Learning Force Fields with Differentiable Boltzmann Estimators
1/ This paper introduces StABlE Training, a novel approach to training machine learning force fields (MLFFs) for molecular dynamics (MD) simulations, which enhances stability by integrating quantum mechanical data and system observables.
2/ Unlike traditional MLFF training, which may lead to unstable simulations, StABlE Training uses Boltzmann Estimators to efficiently guide the model, identifying and correcting unstable regions in molecular phase space during MD simulations.
3/ The method operates in alternating simulation and learning phases. In the simulation phase, multiple MD replicas are run in parallel to identify instability, while the learning phase uses observable data to fine-tune the model, improving its stability without the need for additional quantum mechanical calculations.
4/ A key advantage of StABlE Training is that it requires no extra ab-initio calculations for model correction, making it computationally efficient. It can refine the model using reference observables such as radial distribution functions, diffusivity coefficients, and bond lengths.
5/ The method has been demonstrated on several systems, including organic molecules, tetrapeptides, and water, significantly improving the stability of MLFF simulations, recovering key simulation observables more accurately than traditional methods.
6/ Notably, StABlE-trained models allow for larger time steps in MD simulations without compromising stability, which is crucial for studying long-timescale phenomena like protein folding or ion diffusion.
7/ Results show that StABlE Training not only produces more stable simulations but also outperforms models trained with up to 50 times more quantum mechanical data, making it a highly efficient method for training reliable force fields.
8/ This work paves the way for using MLFFs in more complex and large-scale simulations, expanding the applicability of machine learning in atomistic simulations while minimizing the computational cost.
💻Code: github.com/ASK-Berkeley/StAB…
📜Paper: arxiv.org/abs/2402.13984
#MachineLearning #MolecularDynamics #ForceFields #AI #ComputationalChemistry #QuantumMechanics #DataScience #MDsimulation
1
940
1 Mar 2025
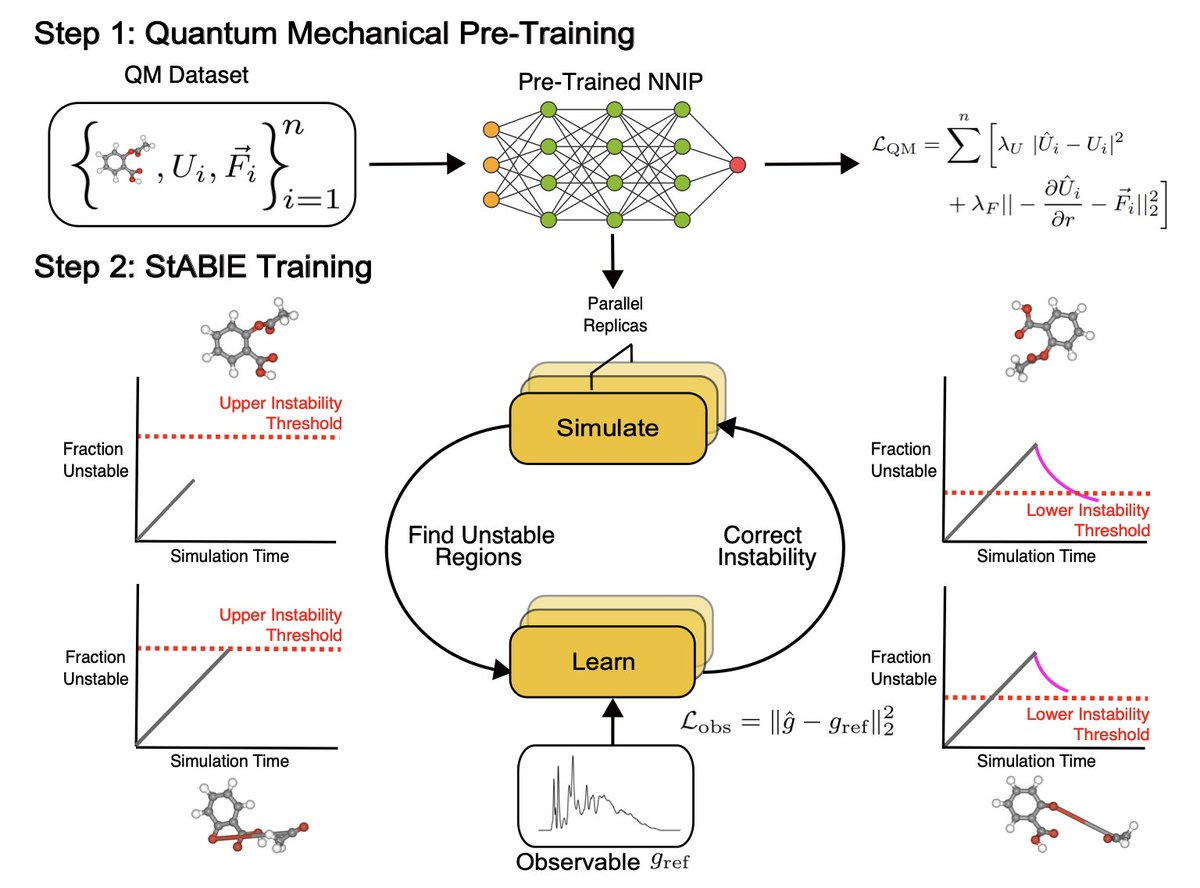
Stability-Aware Training of Machine Learning Force Fields with Differentiable Boltzmann Estimators
1/ This paper introduces StABlE Training, a novel approach to training machine learning force fields (MLFFs) for molecular dynamics (MD) simulations, which enhances stability by integrating quantum mechanical data and system observables.
2/ Unlike traditional MLFF training, which may lead to unstable simulations, StABlE Training uses Boltzmann Estimators to efficiently guide the model, identifying and correcting unstable regions in molecular phase space during MD simulations.
3/ The method operates in alternating simulation and learning phases. In the simulation phase, multiple MD replicas are run in parallel to identify instability, while the learning phase uses observable data to fine-tune the model, improving its stability without the need for additional quantum mechanical calculations.
4/ A key advantage of StABlE Training is that it requires no extra ab-initio calculations for model correction, making it computationally efficient. It can refine the model using reference observables such as radial distribution functions, diffusivity coefficients, and bond lengths.
5/ The method has been demonstrated on several systems, including organic molecules, tetrapeptides, and water, significantly improving the stability of MLFF simulations, recovering key simulation observables more accurately than traditional methods.
6/ Notably, StABlE-trained models allow for larger time steps in MD simulations without compromising stability, which is crucial for studying long-timescale phenomena like protein folding or ion diffusion.
7/ Results show that StABlE Training not only produces more stable simulations but also outperforms models trained with up to 50 times more quantum mechanical data, making it a highly efficient method for training reliable force fields.
8/ This work paves the way for using MLFFs in more complex and large-scale simulations, expanding the applicability of machine learning in atomistic simulations while minimizing the computational cost.
@ask1729 @fpedregosa
💻Code: github.com/ASK-Berkeley/StAB…
📜Paper: arxiv.org/abs/2402.13984
#MachineLearning #MolecularDynamics #ForceFields #AI #ComputationalChemistry #QuantumMechanics #DataScience #MDsimulation
2
1
6
1,233