
We just wrapped up a hands-on scRNA-seq data analysis workshop for PhD students at @DKFZ 🎉
From experimental design to biological interpretation, researchers worked through the complete single-cell analysis journey.
#scRNAseq #SingleCellRNAseq #Bioinformatics #

1
1
25

Jun 7
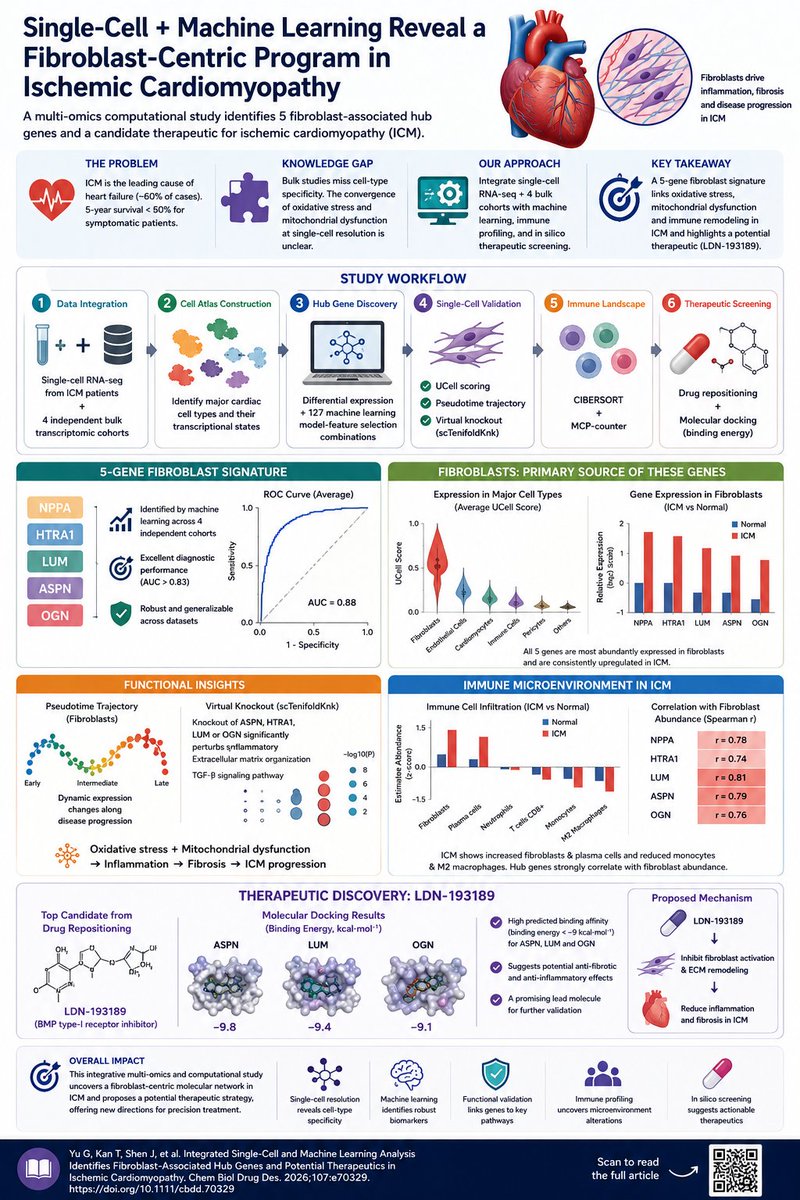
❤️ Single-Cell Machine Learning Reveal a Fibroblast-Centric Program in Ischemic Cardiomyopathy
Why do some hearts continue to deteriorate long after the initial ischemic injury?
A new study in Chemical Biology & Drug Design combined single-cell RNA sequencing, machine learning, immune deconvolution, pseudotime analysis, virtual gene knockout, and molecular docking to uncover the cellular programs driving ischemic cardiomyopathy (ICM).
The investigators integrated scRNA-seq data with four independent transcriptomic cohorts, creating a high-resolution cellular atlas of human ICM. Among 127 machine-learning model combinations, a robust 5-gene diagnostic signature emerged:
🧬 NPPA
🧬 HTRA1
🧬 LUM
🧬 ASPN
🧬 OGN
These genes consistently achieved strong diagnostic performance across independent datasets (AUC > 0.83).
The most striking finding?
All five genes were predominantly expressed in cardiac fibroblasts, identifying fibroblasts as a central orchestrator of ischemic remodeling. Single-cell analyses showed these genes were consistently upregulated in ICM hearts, particularly within fibroblast populations.
Functional analyses pointed toward a shared biological theme:
⚡ Oxidative stress
⚡ Mitochondrial dysfunction
⚡ Extracellular matrix remodeling
⚡ TGF-β signaling
⚡ Inflammatory regulation
Virtual knockout experiments revealed that disruption of ASPN, HTRA1, LUM, or OGN consistently perturbed inflammatory-response pathways, highlighting a fibroblast-driven inflammatory network that may fuel disease progression.
Immune profiling added another layer.
ICM samples showed:
⬆️ Increased fibroblast infiltration
⬆️ Increased plasma cells
⬇️ Reduced monocytes and M2 macrophages
Moreover, all five hub genes strongly correlated with fibroblast abundance, linking fibrosis and immune remodeling into a unified disease program.
The therapeutic angle is particularly interesting.
Computational drug repositioning identified LDN-193189, a BMP type-I receptor inhibitor, as the top candidate. Molecular docking predicted strong binding to ASPN, LUM, and OGN, with binding energies below −9 kcal/mol, suggesting potential anti-fibrotic activity in ICM.
Why this matters
Heart failure research has traditionally focused on cardiomyocytes. This study shifts attention toward fibroblast-centered inflammatory remodeling, suggesting that fibroblasts are not merely scar-forming cells but active regulators of oxidative stress, immune signaling, and disease progression.
The combination of single-cell biology, machine learning, and in silico therapeutics provides a blueprint for discovering actionable targets in complex cardiovascular diseases.
Reference
Yu G, Kan T, Shen J, et al. Integrated Single-Cell and Machine Learning Analysis Identifies Fibroblast-Associated Hub Genes and Potential Therapeutics in Ischemic Cardiomyopathy. Chemical Biology & Drug Design (2026). DOI: 10.1111/cbdd.70329.
#Cardiology #HeartFailure #IschemicCardiomyopathy #SingleCellRNAseq #MachineLearning #Fibroblasts #CardiacFibrosis #SystemsBiology #DrugDiscovery #PrecisionMedicine #Bioinformatics #CardioResearch
4
12
692

May 31
⏳ Early Bird Discount Ends Today!
Learn how real scRNA-seq analysis works — from raw data to biological discovery — in our 3-day hands-on workshop.
🔗 Register: omicsnexus.org/
#scRNAseq #Bioinformatics #SingleCellRNASeq #OmicsNexus

2
2
22
May 30
⏳ 3 days left for the Early Bird Discount!
Learn how real scRNA-seq analysis works — from raw data to biological discovery — in our 3-day hands-on workshop.
🔗 Register: omicsnexus.org/
#scRNAseq #Bioinformatics #SingleCellRNASeq #OmicsNexus

2
2
5

May 27
🔬 Planning your next single-cell project? Take advantage of reduced pricing on Active Motif’s Single-Cell RNA-Seq services for new projects today!
Request an epigenetics quote today: bit.ly/49jPLjM
______
#SingleCell #SingleCellRNASeq #LifeScience #Biotech #RNASeq

1
3
147

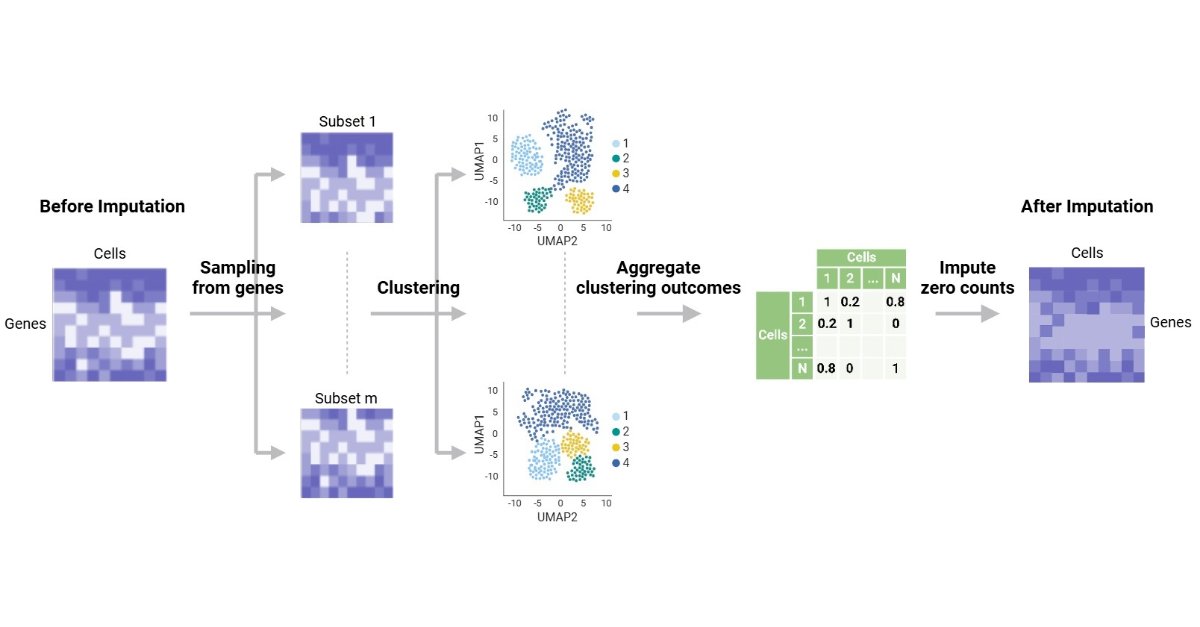
💥Excited for the publication: "CCI: A Consensus Clustering-Based Imputation Method for Addressing Dropout Events in scRNA-Seq Data"
🔗 brnw.ch/21x1ABk
📌 #SingleCellRNAseq #scRNAseq #Bioinformatics #DataImputation #Genomics #MachineLearning #ComputationalBiology
2
3
81

Apr 10
🚨𝟒-𝐃𝐚𝐲 𝐇𝐚𝐧𝐝𝐬-𝐎𝐧 𝐎𝐧𝐥𝐢𝐧𝐞 𝐖𝐨𝐫𝐤𝐬𝐡𝐨𝐩 𝐨𝐧 𝐒𝐢𝐧𝐠𝐥𝐞-𝐂𝐞𝐥𝐥 𝐑𝐍𝐀-𝐒𝐞𝐪 𝐃𝐚𝐭𝐚 𝐀𝐧𝐚𝐥𝐲𝐬𝐢𝐬! 🧬💻
🔗 𝐑𝐞𝐠𝐢𝐬𝐭𝐞𝐫 𝐡𝐞𝐫𝐞: forms.gle/peZsgkNySJ2Wqf2UA
#SingleCellRNASeq #scRNAseq #TrajectoryAnalysis #CellCellCommunication

1
1
34
Benchmarking Algorithms for RNA Velocity Inference
1. A comprehensive study benchmarks 29 RNA velocity inference algorithms across 176 datasets, including simulated and real scRNA-seq data. This work provides a systematic evaluation framework focusing on accuracy, scalability, stability, and usability. The results highlight that no single method is universally optimal, emphasizing the importance of selecting tools based on specific data modalities and biological contexts.
2. The study introduces a unified benchmarking framework that decomposes performance into four practical dimensions. Accuracy is assessed through metrics like vector correlation, clustering coherence, and spatial consistency. Scalability is evaluated by runtime and memory usage across datasets of varying sizes. Stability tests include robustness to input perturbations and reproducibility across multiple runs.
3. Key findings include veloVI ranking as the top overall method due to its balanced performance across metrics. The study also identifies challenges such as scalability limitations for large datasets, sensitivity to gene selection, and the lack of truly multimodal and spatially explicit velocity models. Practical guidelines are provided for selecting RNA velocity tools based on data type, available priors, and computational constraints.
4. For spatial and multi-omics data, the study evaluates several specialized methods. Spatial velocity methods like SDEvelo and STT show robust performance in capturing spatially coherent dynamics, while multi-omics approaches like scKINETICS and MultiVelo demonstrate the potential for integrating additional modalities to improve velocity inference.
5. The study concludes that RNA velocity is a powerful tool for visualizing cellular dynamics but requires careful consideration of its limitations. Future development should focus on integrating multimodal data and improving scalability to handle large-scale datasets.
📜Paper: biorxiv.org/content/10.64898…
#RNAVelocity #SingleCellRNASeq #Benchmarking #ComputationalBiology #Multiomics #SpatialTranscriptomics

1
3
14
1,366

16 Dec 2025
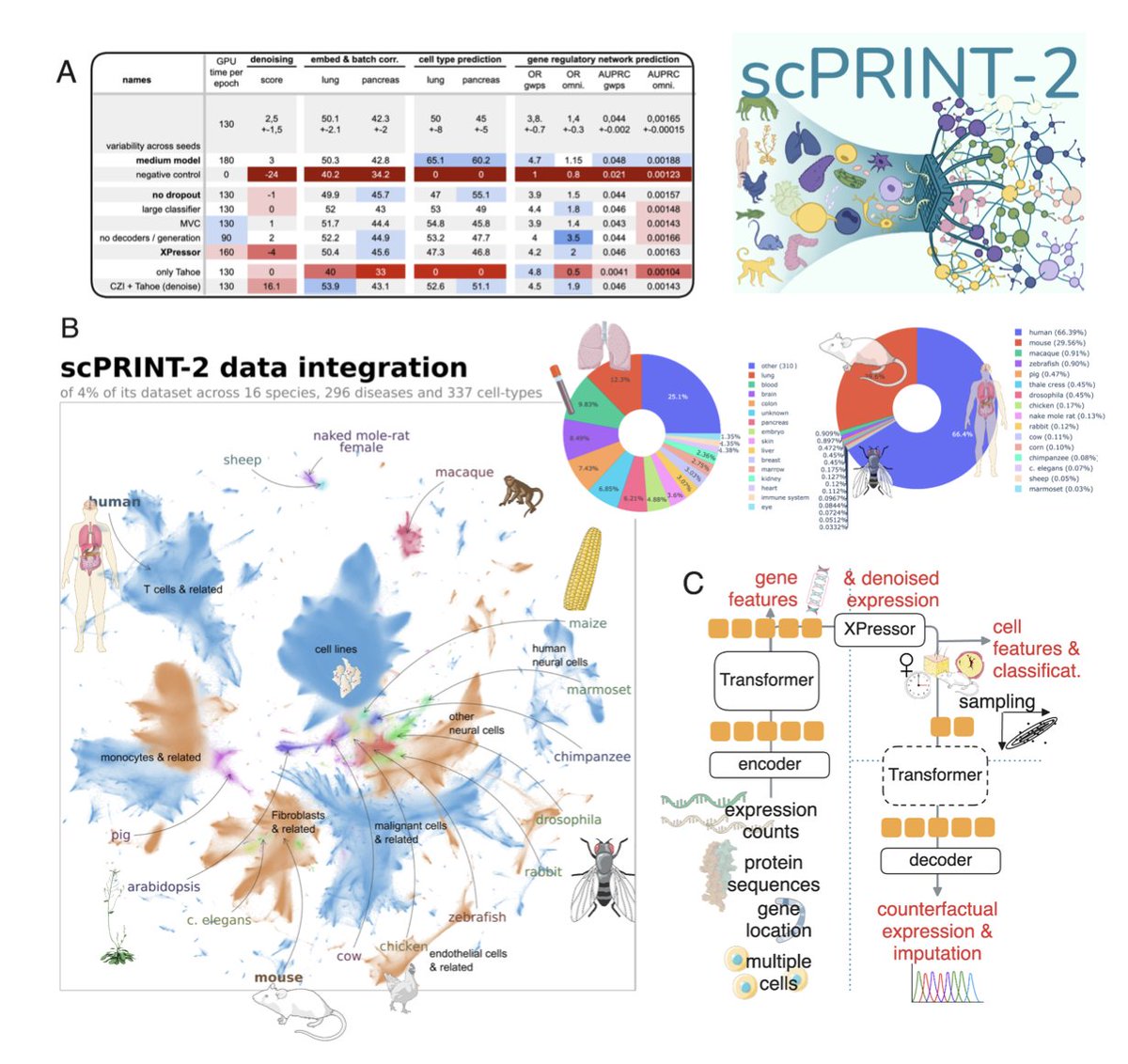
ScPRINT-2: Towards the Next-Generation of Cell Foundation Models and Benchmarks
1. A novel preprint introduces scPRINT-2, a next-generation single-cell foundation model pre-trained on an unprecedented scale of 350 million cells across 16 organisms. This model pushes the boundaries of single-cell RNA-seq analysis by achieving state-of-the-art performance in tasks like expression denoising, cell embedding, and cell type prediction.
2. The study presents an additive benchmark across a diverse set of tasks to systematically evaluate the impact of different model architectures, pre-training datasets, and training modalities. This comprehensive approach helps identify key features that enhance model performance and generalization capabilities.
3. scPRINT-2 incorporates several innovative components, including a graph neural network encoder, XPressor architecture, and a novel loss function combining zero-inflated negative binomial and mean squared error. These contributions enable the model to handle diverse data types and improve its accuracy in challenging contexts.
4. The model demonstrates remarkable generalization ability, successfully predicting cell types and gene networks in unseen organisms and modalities. This highlights the potential of scPRINT-2 for cross-species analysis and its ability to generate high-quality embeddings and counterfactual reasoning results.
5. The scPRINT-2 corpus, the largest single-cell dataset ever assembled, includes over 400,000 distinct genes and 140,000 cell groups. This rich dataset, combined with advanced data augmentation techniques, plays a crucial role in enhancing the model's performance and versatility.
6. The authors emphasize the importance of open-sourcing not just the model weights but also the pre-training tasks, datasets, and training traces. This transparency and reproducibility set a new standard for the field and enable further advancements in single-cell biology.
📜Paper: biorxiv.org/content/10.64898…
#SingleCellRNASeq #FoundationModels #Bioinformatics #MachineLearning #OpenScience
25
87
5,872
29 Nov 2025
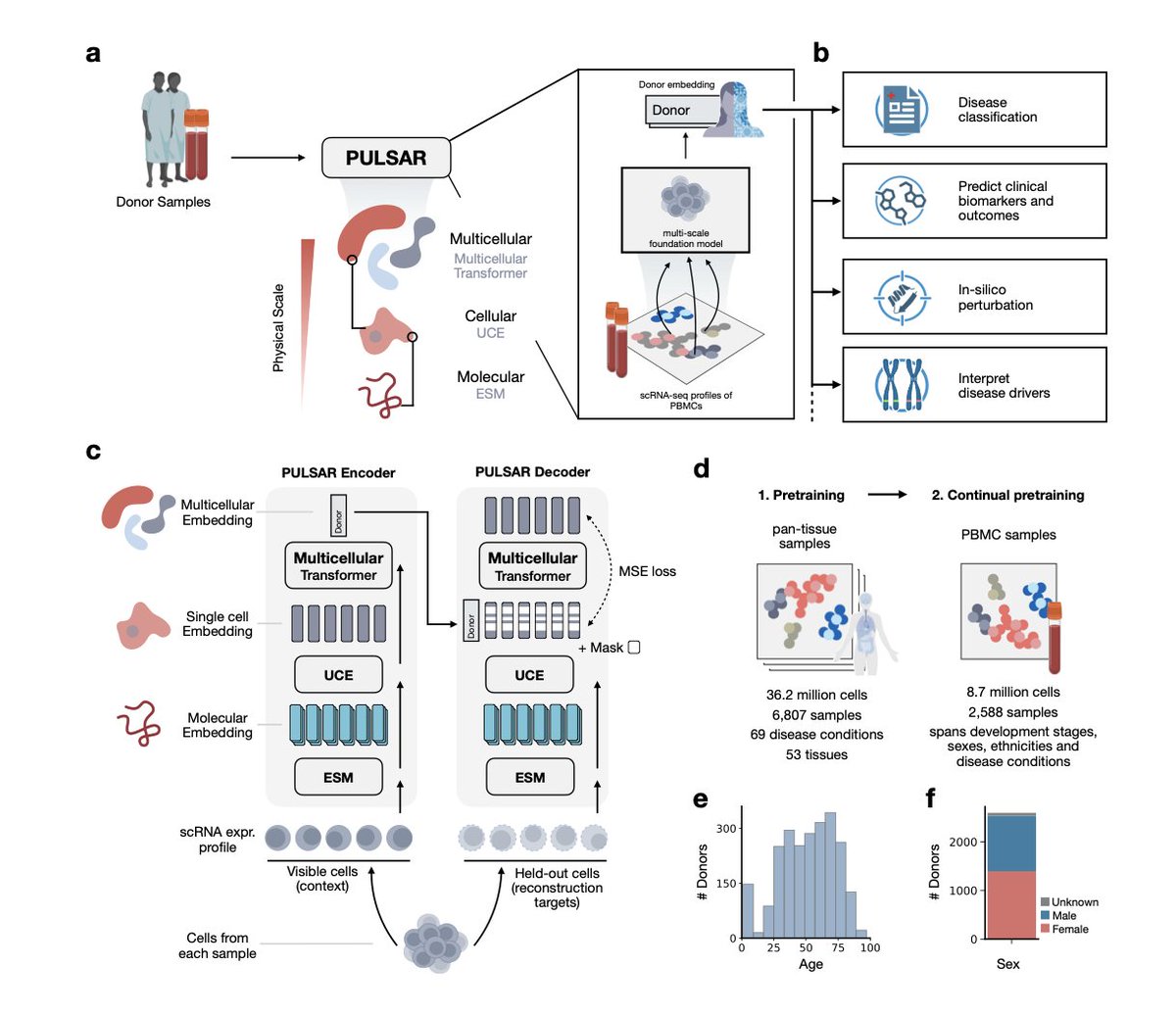
Pulsar: A Foundation Model for Multi-scale and Multi-cellular Biology
1. PULSAR is a groundbreaking multi-scale foundation model that integrates molecular, cellular, and multicellular information to create unified donor representations. This model leverages single-cell RNA sequencing data to capture the complexity of biological systems across different scales, enabling accurate disease classification and prediction of clinical events.
2. A key innovation of PULSAR is its hierarchical architecture, which allows information to flow from genes to cells and then to multicellular systems. This design enables the model to capture emergent properties at the individual level, providing a comprehensive understanding of health and disease states.
3. Applied to the human peripheral immune system, PULSAR demonstrates exceptional performance in disease classification, achieving state-of-the-art accuracy in distinguishing between various inflammatory conditions. The model's ability to generalize across diverse studies and disease conditions highlights its robustness.
4. PULSAR's generative capabilities allow it to simulate cytokine perturbation responses across physical scales, revealing how perturbations at the molecular level impact cellular and multicellular systems. This feature is crucial for understanding the context-dependent effects of cytokines in immune responses.
5. The model's interpretability is a significant advantage, as it identifies key cell types and gene programs driving disease progression. This capability provides valuable insights into the underlying biological mechanisms and could inform therapeutic targeting strategies.
6. PULSAR also shows strong predictive power for future clinical events, such as rheumatoid arthritis onset and flu vaccine responsiveness. These applications highlight the model's potential for precision medicine and personalized healthcare.
7. Despite its achievements, PULSAR faces limitations, such as the lack of spatial context in dissociated single-cell data and the absence of immune receptor repertoires. Future work will focus on incorporating spatial information and additional omics data to enhance the model's capabilities.
📜Paper: biorxiv.org/content/10.1101/…
#ComputationalBiology #MultiScaleModeling #SingleCellRNASeq #PrecisionMedicine #AIinBiology
2
20
1,796
27 Nov 2025
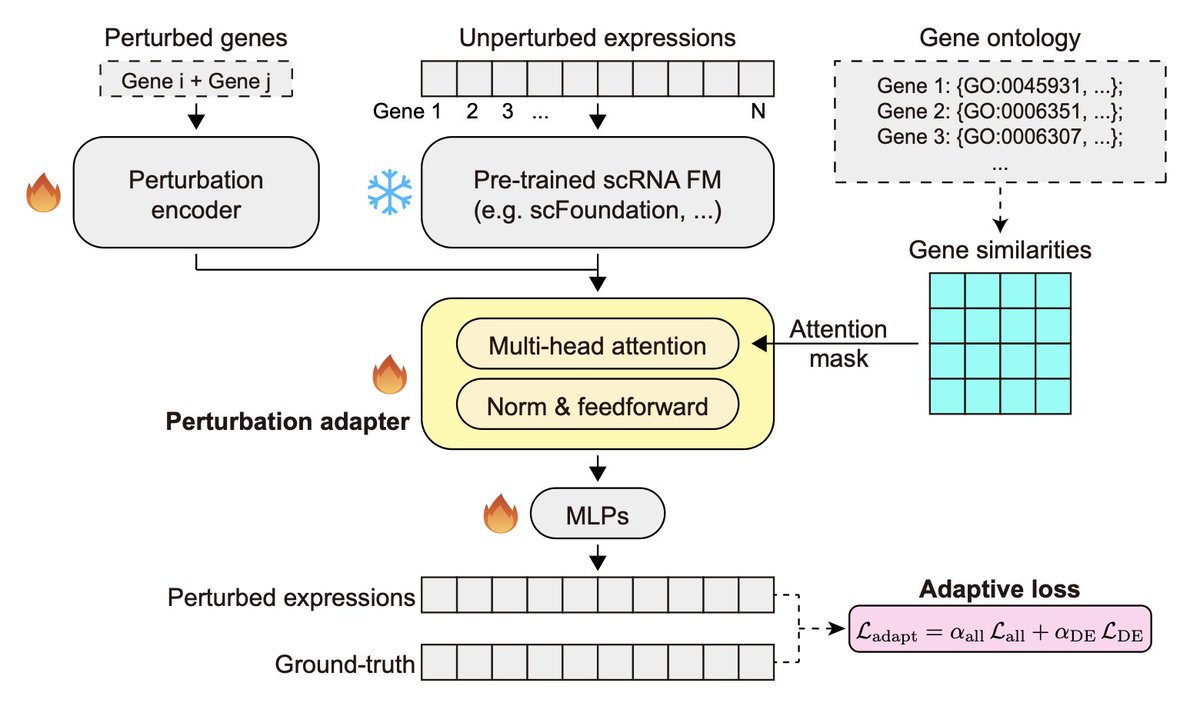
PertAdapt: Unlocking Single-Cell Foundation Models for Genetic Perturbation Prediction via Condition-Sensitive Adaptation
1. A new framework called PertAdapt has been introduced to enhance the prediction of genetic perturbation effects using single-cell foundation models. This method integrates a perturbation adapter and an adaptive loss to improve the accuracy of transcriptional response predictions.
2. The core innovation of PertAdapt is its perturbation adapter, which employs a gene-similarity-masked attention mechanism. This allows the model to effectively transfer knowledge from large-scale pretraining datasets to specific perturbation tasks, addressing the challenge of imbalanced gene responses.
3. The adaptive loss function dynamically reweights perturbation-sensitive genes relative to global transcriptomic signals. This approach ensures that the model prioritizes meaningful differential expression patterns, leading to significant improvements in prediction accuracy.
4. Extensive experiments across seven diverse datasets demonstrate PertAdapt's superior performance compared to both non-pretrained and existing foundation model baselines. The framework shows strong generalization, especially in limited-data regimes and for modeling multiplexed gene interactions.
5. PertAdapt's scalability is highlighted through experiments with different-sized foundation models. The results show that the framework maintains robust performance across varying model capacities, indicating its capacity-insensitive nature.
6. Ablation studies confirm the necessity of each component in PertAdapt, including the pretrained foundation model, the adapter, the attention mask, and the adaptive loss. These components work synergistically to unlock the full potential of single-cell foundation models for perturbation prediction.
📜Paper: biorxiv.org/content/10.1101/…
#PertAdapt #GeneticPerturbation #SingleCellRNASeq #FoundationModels #ComputationalBiology #Bioinformatics
6
813
20 Nov 2025
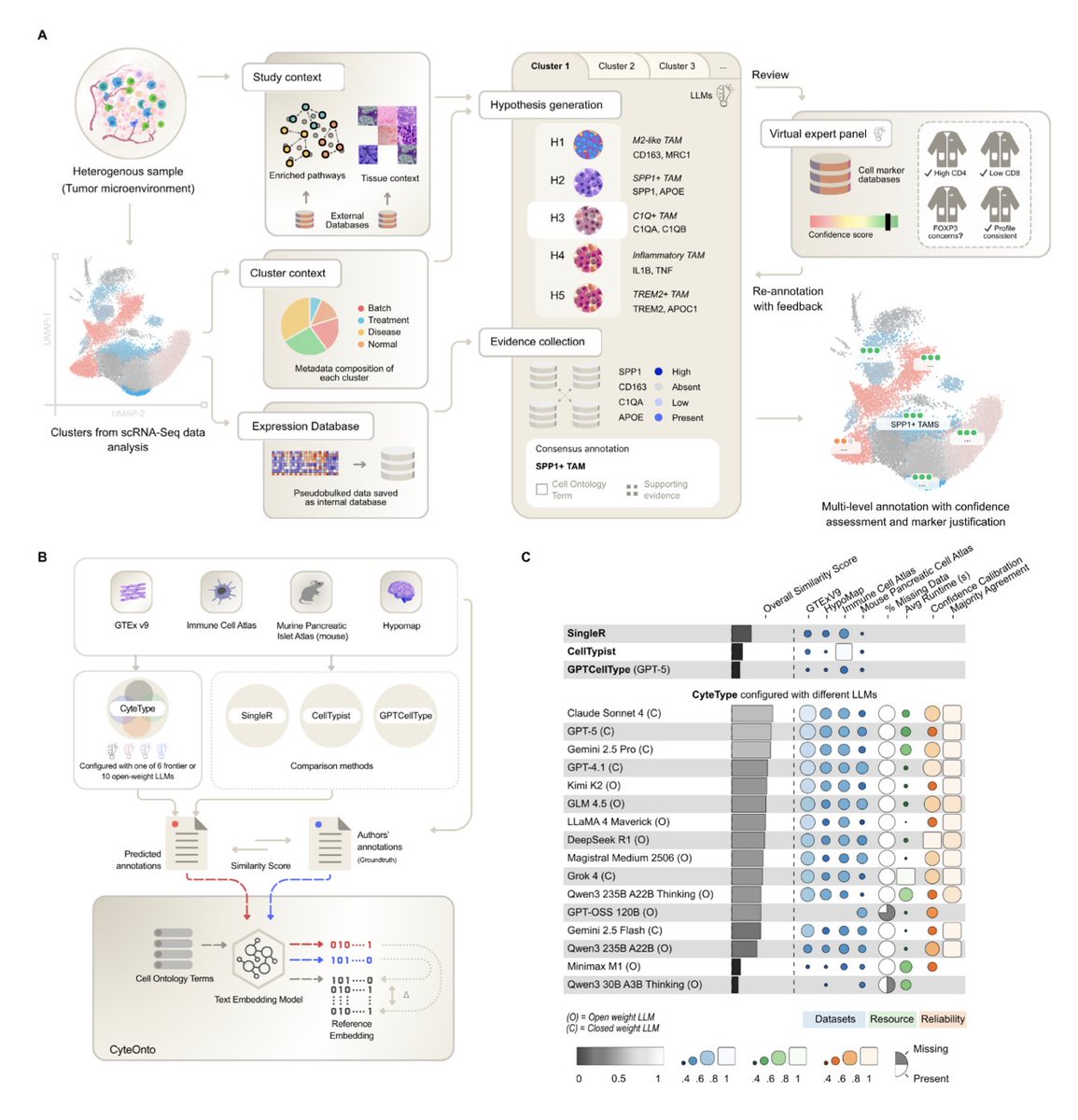
Multi-agent AI enables evidence-based cell annotation in single-cell transcriptomics
1. A new multi-agent framework called CyteType has been developed to address the critical bottleneck of cell type annotation in single-cell transcriptomics. This framework leverages full expression data and study context to generate competing hypotheses, validate predictions against external databases, and iteratively self-evaluate.
2. CyteType outperforms both reference-based and LLM-based methods in comprehensive benchmarking. It provides self-generated confidence scores that reliably identify trustworthy annotations, transforming cell type annotation from a simple label assignment into an evidence-grounded biological discovery process.
3. The framework includes a novel semantic similarity framework called CyteOnto, which maps cell type labels to Cell Ontology terms using text embeddings. This approach captures semantic relationships more effectively than traditional methods, enabling quantitative comparison of annotations.
4. CyteType’s architecture is robust across diverse LLMs, including both closed-weight and open-weight models. It demonstrates significant performance gains over existing methods, with high-confidence annotations showing higher similarity scores and heterogeneous clusters flagged for targeted expert review.
5. Applying CyteType to 977 clusters across 20 datasets revealed that it not only validates existing annotations but also adds functional enhancements, refines subtypes, and identifies clusters requiring major reannotation, highlighting its potential for novel discoveries.
6. CyteType is available as Python and R packages, integrating seamlessly into existing workflows. It generates comprehensive reports with interactive HTML, providing detailed marker justification, pathway enrichment, literature evidence, and disease associations.
7. This study demonstrates that CyteType’s multi-agent architecture and structured reasoning significantly improve upon direct LLM prompting, offering a transparent and interpretable process for cell type annotation with quantified uncertainty.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/NygenAnalytics/Cy…
#SingleCellRNASeq #CellAnnotation #AI #Bioinformatics #MultiAgentSystems #CyteType
1
1
5
730
17 Nov 2025
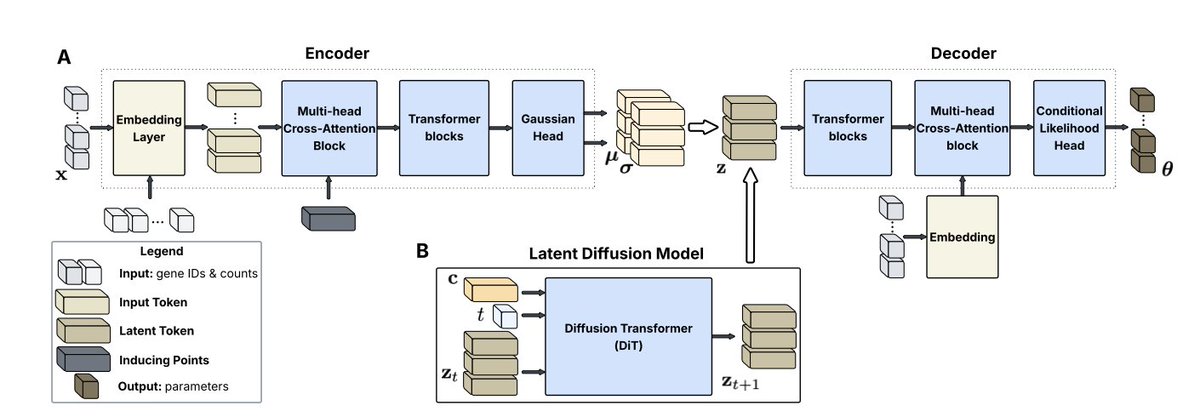
Scalable Single-Cell Gene Expression Generation with Latent Diffusion Models
1. This paper introduces scLDM, a novel latent diffusion model for generating single-cell gene expression profiles. It addresses key challenges in existing models, such as artificial gene orderings and training instabilities, by leveraging a fully transformer-based architecture and a latent diffusion framework.
2. The core innovation lies in the use of a Multi-head Cross-Attention Block (MCAB) that serves dual purposes: permutation-invariant pooling in the encoder and permutation-equivariant unpooling in the decoder. This design respects the exchangeable nature of gene expression data, allowing for high-quality and scalable generation.
3. scLDM replaces the standard Gaussian prior with a latent diffusion model using Diffusion Transformers and linear interpolants. This enables controlled generation through classifier-free guidance, allowing the model to generate cell states conditioned on multiple attributes simultaneously.
4. The model demonstrates superior performance in reconstructing and generating single-cell data across various benchmark datasets. It achieves higher Pearson correlation coefficients and lower reconstruction errors compared to existing methods like scVI and CFGen.
5. In conditional generation tasks, scLDM shows its ability to generate realistic cell states under different perturbations and cell contexts. This is crucial for understanding cellular responses to genetic and chemical perturbations.
6. The embeddings learned by scLDM's VAE also prove useful in downstream classification tasks, such as identifying infected cells in COVID-19 datasets and classifying cell types in Tabula Sapiens 2.0. This highlights the model's potential for biological discovery.
7. The authors provide detailed experiments and ablation studies, demonstrating the model's robustness and the impact of different architectural choices. They also compare different classifier-free guidance strategies, showing that joint conditioning outperforms additive conditioning in multi-attribute settings.
💻Code: github.com/czi-ai/scLDM
📜Paper: arxiv.org/abs/2511.02986
#SingleCellRNASeq #LatentDiffusion #GenerativeModeling #ComputationalBiology #AIinBiology
1
8
1,124
7 Nov 2025
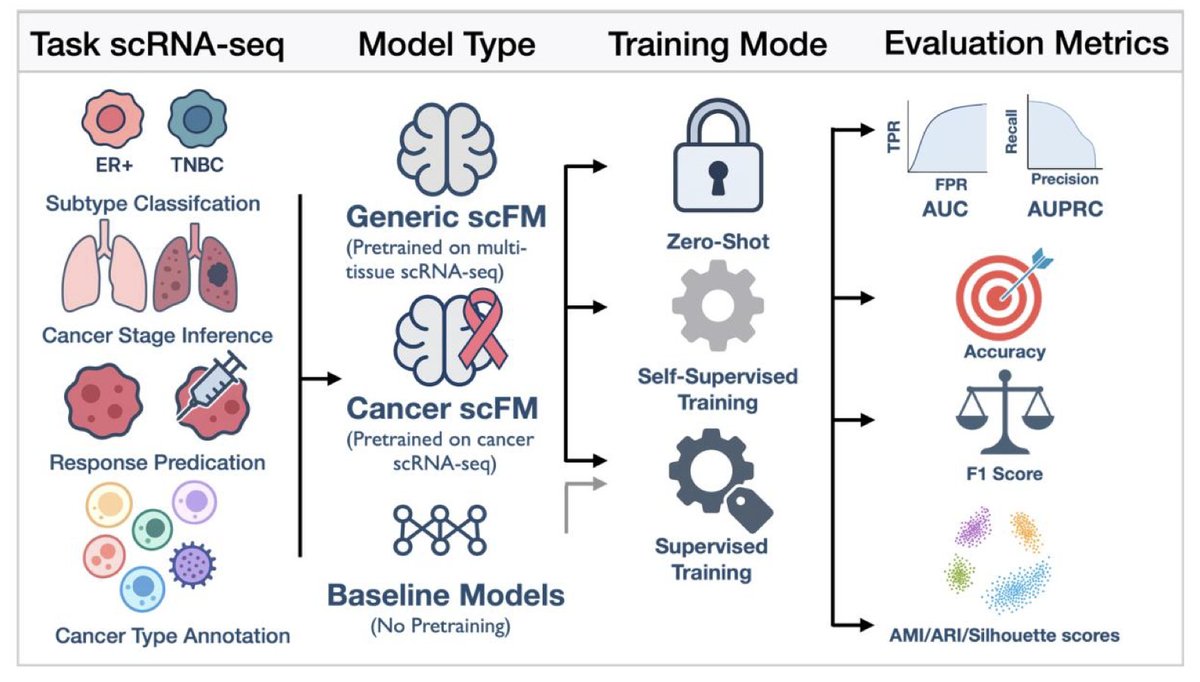
Empirical Evaluation of Single-Cell Foundation Models for Predicting Cancer Outcomes
1. This study systematically evaluates nine single-cell foundation models (scFMs) and three baseline approaches across six cancer-specific tasks, revealing their potential and limitations in predicting clinical outcomes.
2. The research highlights that while scFMs excel in tumor microenvironment cell annotation, they show limited advantages in predicting patient-level outcomes compared to simpler baseline models.
3. A key finding is that continual training and domain adaptation significantly enhance the performance of scFMs, even outperforming larger models in some cases.
4. The study explores various patient-level aggregation strategies, finding that multi-instance learning (MIL) generally leads to the highest performance, though with higher variability.
5. End-to-end fine-tuning did not consistently outperform strong baseline methods, suggesting that careful model adaptation is crucial for clinical applications.
6. The authors emphasize the need for further methodological innovation and larger cancer cohorts to improve the translational impact of scFMs in precision oncology.
7. The study underscores the importance of evaluating scFMs on clinically relevant tasks to guide future model development and ensure their applicability in real-world settings.
📜Paper: biorxiv.org/content/10.1101/…
#SingleCellRNASeq #FoundationModels #CancerResearch #PrecisionOncology #Bioinformatics
3
872
5 Nov 2025
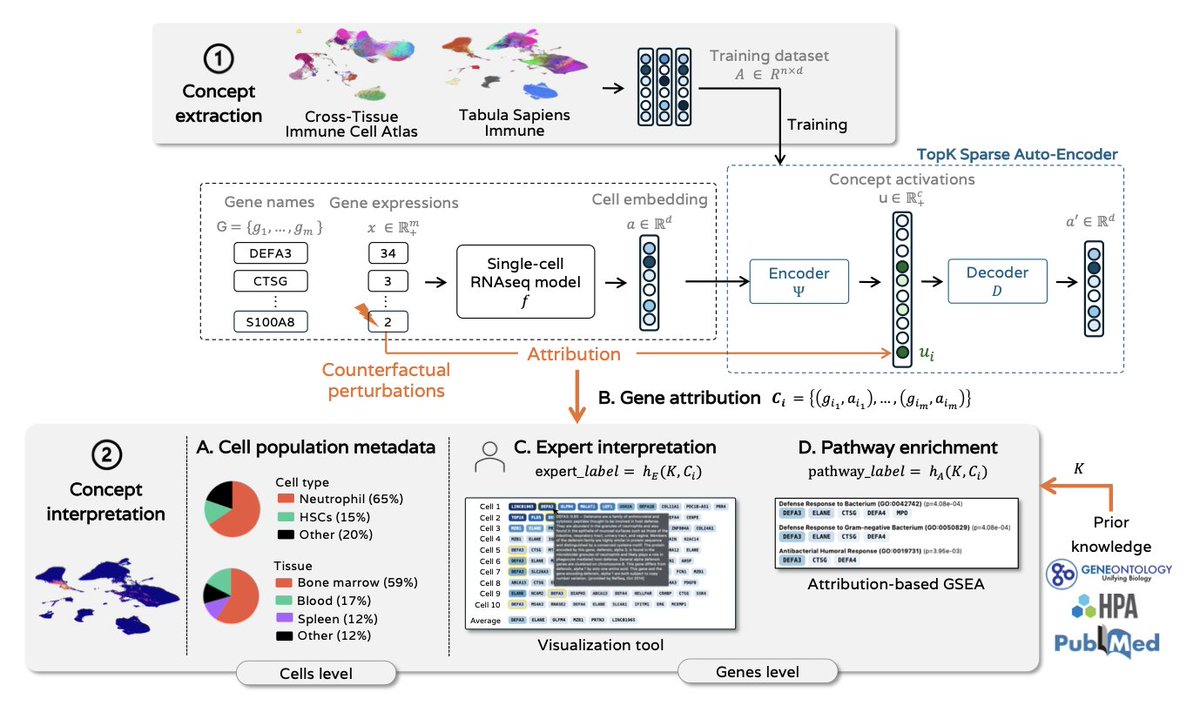
Discovering Interpretable Biological Concepts in Single-Cell RNA-Seq Foundation Models
1. This study introduces a novel framework for interpreting biological concepts in single-cell RNA-seq models using sparse autoencoders, addressing the challenge of making deep learning models more transparent and useful for biological discovery.
2. The researchers propose an attribution method with counterfactual perturbations to identify genes that influence concept activation, moving beyond traditional correlational approaches like differential gene expression analysis.
3. Two complementary interpretation approaches are provided: an expert-driven analysis facilitated by an interactive interface and an ontology-driven method using attribution-based biological pathway enrichment.
4. Applying this framework to well-known single-cell RNA-seq models, the study demonstrates that concepts extracted by sparse autoencoders are more interpretable than individual neurons and preserve the richness of latent representations.
5. The findings show that concepts improve interpretability compared to individual neurons, aligning well with biological signals such as cell types and biological processes, and reveal a set of stable concepts across datasets.
6. The work also demonstrates that concept activations preserve predictive performance in downstream tasks like cell type and cell cycle phase classification, making the concept space useful for interpretable analyses.
📜Paper: arxiv.org/abs/2510.25807
#SingleCellRNASeq #DeepLearning #BiologicalConcepts #Interpretability #ComputationalBiology
1
5
1,017
26 Oct 2025
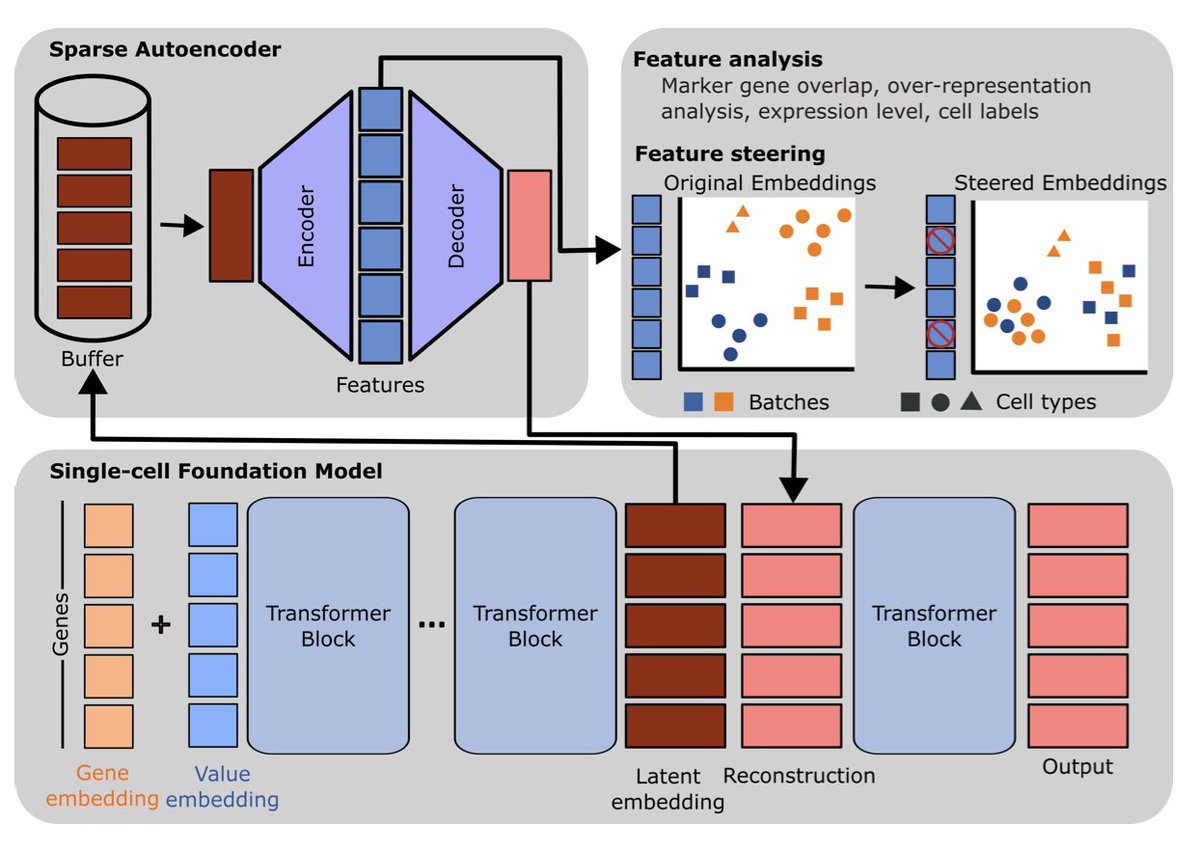
Sparse Autoencoders Reveal Interpretable Features in Single-Cell Foundation Models
1. A new study by Pedrocchi et al. explores how sparse autoencoders (SAEs) can uncover meaningful biological and technical signals in single-cell foundation models (scFMs). This work provides insights into the complex representations learned by scFMs, which are crucial for applications like cell type annotation and data integration.
2. The researchers trained SAEs on the hidden representations of two widely used scFMs, scGPT and scFoundation. They found that these models capture diverse biological concepts, even in their pre-trained states, revealing both gene-specific and cell-specific features.
3. An important finding is that different training protocols and architectures of scFMs lead to distinct encodings of information. This suggests that the internal mechanisms of scFMs are highly influenced by their training procedures.
4. The study also highlights that while scFMs can capture cell type information across multiple studies, they often fail to unify this information into a single generalized representation. This limitation affects the models' generalizability across different datasets.
5. A novel contribution is the demonstration that SAE-derived features are causally related to model behavior. The authors show that by manipulating these features, they can reduce unwanted technical effects and improve batch integration in scFMs.
6. The work provides a path toward more interpretable and controllable single-cell foundation models. By understanding and steering the internal representations, researchers can potentially enhance the performance and usability of these models in various biological applications.
📜Paper: biorxiv.org/content/10.1101/…
#SingleCellRNASeq #SparseAutoencoders #InterpretableAI #ComputationalBiology #FoundationModels
1
4
19
1,722
20 Oct 2025
GeneJepa: A Predictive World Model of the Transcriptome
1. GeneJepa introduces a novel self-supervised learning approach for single-cell transcriptomes, shifting from reconstructing raw expression values to predicting latent representations of masked gene sets. This innovative method focuses on capturing the underlying rules of gene regulation rather than just fitting to noisy data.
2. The model employs a Perceiver encoder to handle variable-length gene sets efficiently, allowing for scalable processing of large datasets. A Fourier-feature tokenizer is used to jointly represent gene identity and continuous expression values, preserving quantitative information without binning.
3. GeneJepa demonstrates superior performance in downstream tasks such as cell-type identification and drug response prediction. It outperforms existing models like scGPT, showing robustness across diverse tissues and experimental conditions.
4. A key innovation is the ability to perform zero-shot in-silico gene knockouts by manipulating embeddings with pre-computed perturbation vectors. This suggests that GeneJepa can simulate the effects of genetic interventions without specific training on perturbation pairs.
5. The model’s architecture supports test-time scaling, enabling practitioners to trade off compute for higher accuracy at inference without retraining. This flexibility is particularly useful for applications requiring different levels of precision and resource allocation.
6. Trained on the Tahoe-100M dataset, GeneJepa learns general representations that transfer across tissues and datasets. This large-scale pretraining drives the model to capture causal regularities that generalize well.
📜Paper: biorxiv.org/content/10.1101/…
#GeneJepa #Transcriptome #PredictiveModeling #SingleCellRNASeq #ComputationalBiology #AIinBiology
4
22
1,492
6 Oct 2025
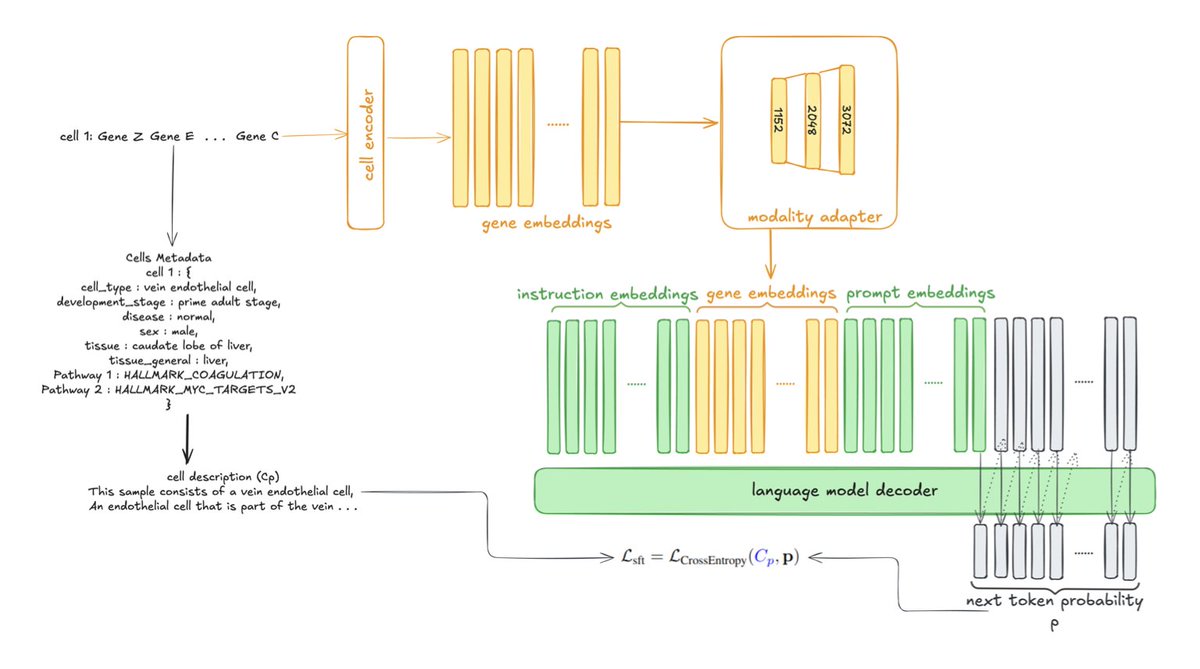
Cell2Text: Multimodal LLM for Generating Single-Cell Descriptions from RNA-Seq Data
1. Cell2Text introduces a novel multimodal generative framework that translates single-cell RNA sequencing profiles into structured natural language descriptions, offering a richer and more interpretable way to understand cellular identity and function compared to traditional classification methods.
2. The model combines gene-level embeddings from the Geneformer, a powerful single-cell foundation model, with pretrained large language models. This integration allows Cell2Text to generate coherent summaries that capture not only cellular identity but also tissue origin, disease associations, and pathway activity.
3. A key innovation is the use of a lightweight adapter module to bridge the gap between the high-dimensional cellular representations from Geneformer and the semantic space of the language model, enabling the generation of detailed and biologically meaningful descriptions.
4. Empirically, Cell2Text demonstrates superior performance in cell type, tissue, and disease classification tasks, outperforming specialized baselines. It also shows strong ontological consistency using PageRank-based similarity metrics, indicating that even incorrect predictions maintain high biological relevance.
5. The model achieves high semantic fidelity in text generation, with BioBERT F1-scores exceeding 93%, showcasing its ability to produce scientifically meaningful and interpretable cellular characterizations.
6. A large-scale multimodal dataset of 1 million cells from CELLxGENE was constructed, enriched with ontology terms, tissue and disease metadata, and pathway annotations. This dataset supports cross-modal training and evaluation at an unprecedented scale.
7. The study highlights the potential of coupling expression data with natural language, pointing to a scalable path for label-efficient characterization of unseen cells and offering a general strategy for building interpretable frameworks in computational biology.
📜Paper: arxiv.org/abs/2509.24840
#Cell2Text #MultimodalLLM #SingleCellRNASeq #NaturalLanguageProcessing #ComputationalBiology
1
2
816
22 Sep 2025
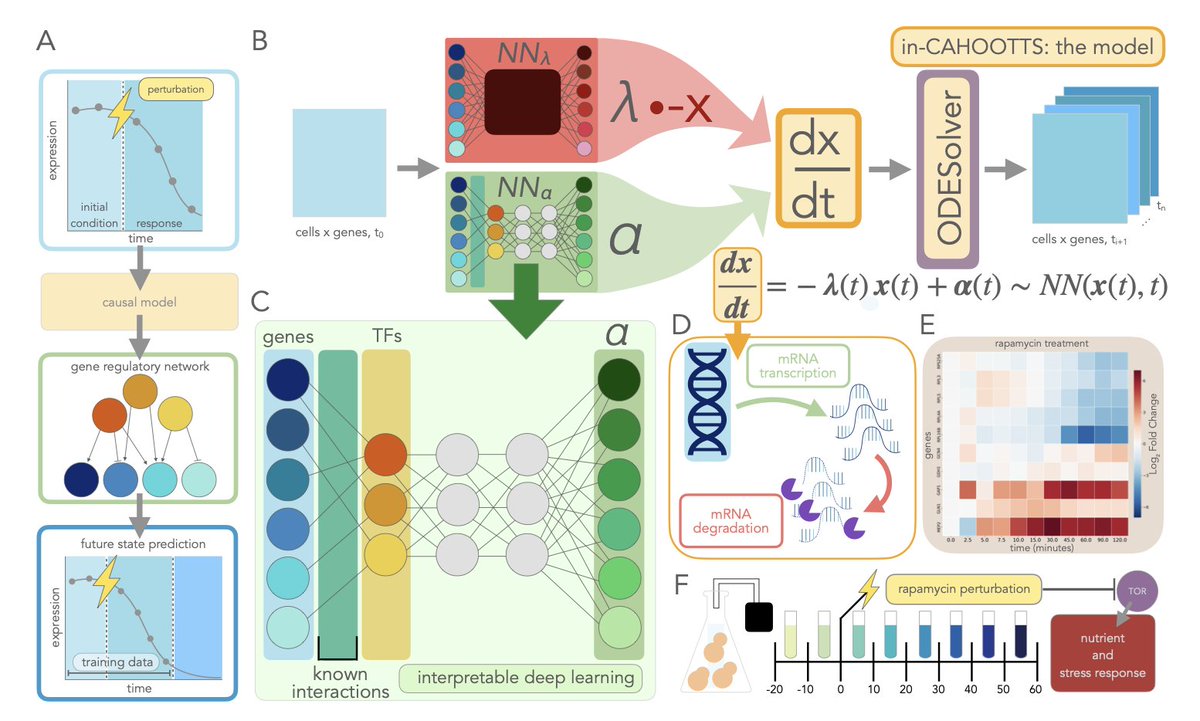
Dynamic Gene Regulatory Network Inference with Interpretable, Biophysically-Motivated Neural ODEs
1. A novel framework called in-CAHOOTTS leverages neural ordinary differential equations (ODEs) to infer gene regulatory networks (GRNs) from dynamic single-cell data, achieving both accurate prediction and mechanistic interpretability.
2. The model decomposes gene expression dynamics into fundamental biophysical processes (mRNA transcription and degradation) while inferring regulatory network structure, validated on yeast responses to rapamycin and cell cycle progression.
3. in-CAHOOTTS demonstrates stable long-term predictions, maintaining realistic oscillatory dynamics for over 40 hours in cell cycle modeling, extending 30 times beyond training data and capturing true biological attractors.
4. The framework incorporates prior biological knowledge as a soft constraint, guiding the model towards established regulatory mechanisms while allowing data-driven discovery of novel interactions, significantly improving GRN inference accuracy.
5. The interpretable architecture reveals biological insights such as the disconnect between latent transcription factor activity and observable mRNA expression, and the underlying GRN driving cellular responses.
6. The model achieves a 60% improvement in predictive accuracy and a 3.5-fold improvement in regulatory network recovery compared to vanilla Neural ODEs, highlighting the importance of biophysical decomposition and prior knowledge integration.
7. The approach is validated on both rapamycin response and cell cycle dynamics, demonstrating its broad applicability and potential for advancing biological understanding through interpretable deep learning.
📜Paper: biorxiv.org/content/10.1101/…
#GeneRegulatoryNetworks #NeuralODEs #SingleCellRNASeq #Biophysics #DeepLearning #SystemsBiology
1
6
28
1,501
20 Sep 2025
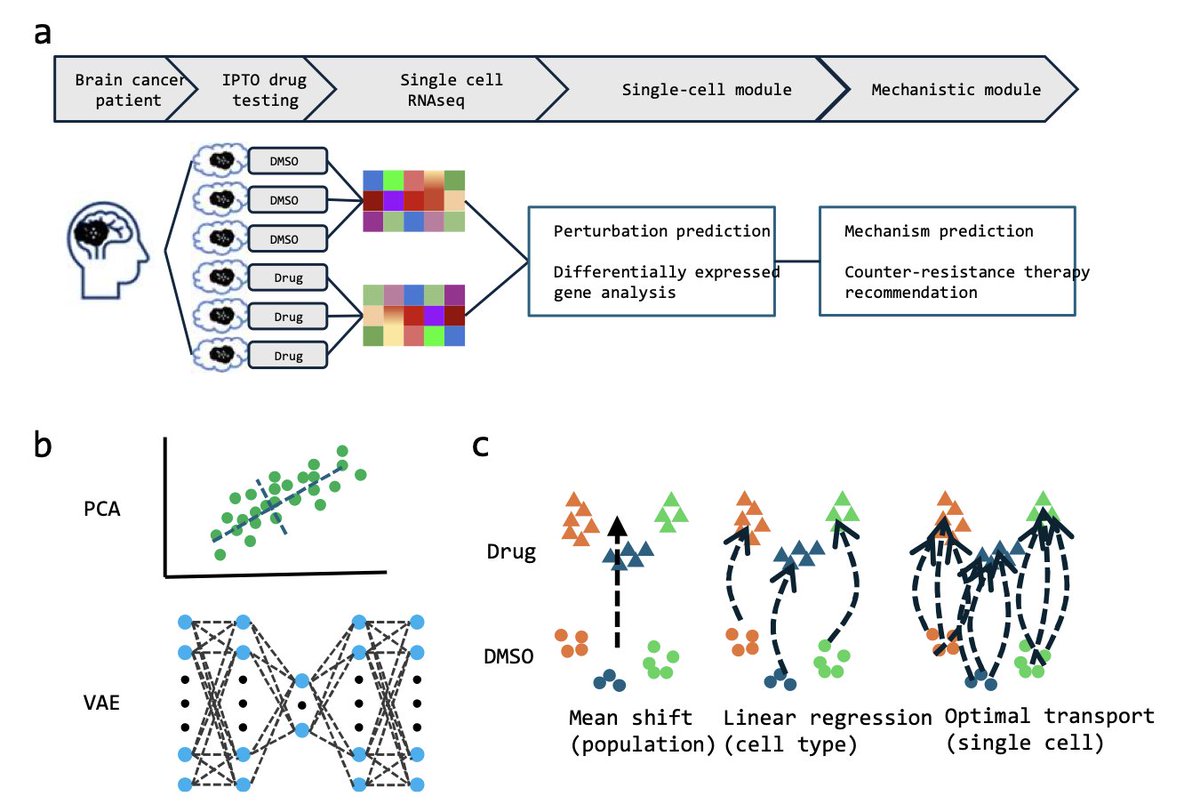
DELPHAI,AI Agent for Predicting Drug Response and Resistance
1. DELPHAI is a novel AI agent that integrates single-cell perturbation prediction with mechanistic analysis using large language models to predict drug responses and resistance mechanisms in patient-derived organoids.
2. The study demonstrates that DELPHAI can accurately predict cellular responses to drug treatment and identify resistance mechanisms without prior knowledge of the drug, showing significant potential for AI-driven precision medicine.
3. DELPHAI uses a comprehensive benchmarking framework that evaluates methods in both computational embedding and reconstructed gene expression spaces, ensuring that computational predictions maintain biological interpretability and clinical relevance.
4. Applied to glioblastoma organoids treated with temozolomide, DELPHAI correctly identified DNA alkylation as the primary mechanism of action and recommended combination therapies that align with established clinical strategies.
5. The AI agent's therapeutic recommendations are strongly validated by clinical literature, including combining TMZ with DNA repair inhibitors and targeting glioma stem cells, demonstrating its ability to translate computational predictions into clinically relevant insights.
6. The study highlights the importance of systematic benchmarking and the use of appropriate evaluation metrics, such as energy distance, to reveal the superior performance of certain methods like optimal transport in capturing complex population dynamics.
7. DELPHAI's approach represents a significant advance toward AI-driven drug discovery and precision medicine in cancer treatment, though further validation across diverse datasets and clinical integration are needed for broader application.
📜Paper: biorxiv.org/content/10.1101/…
#AI #PrecisionMedicine #CancerTherapy #DrugResistance #SingleCellRNASeq #ComputationalBiology
4
22
1,601