
New lecture in a series on Generative Modeling for Control problems. I introduce a control version of gradient descent: The nonholonomic gradient descent.
hackmd.io/pTtk6ohHSBSfqLVsNU…
For other lectures: sites.google.com/view/karthi…
#Controltheory #generativemodeling
2
117
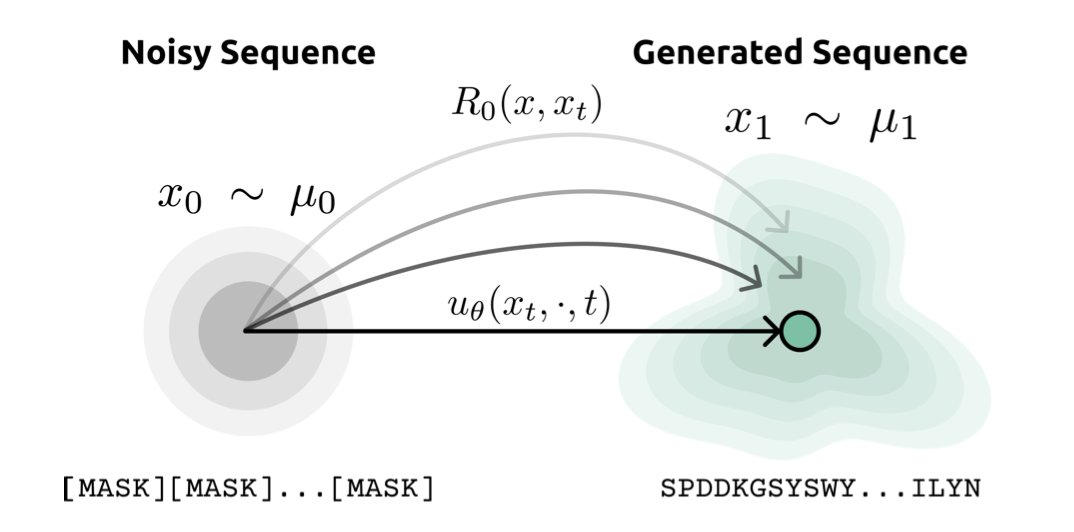
Minimal-Action Discrete Schrödinger Bridge Matching for Peptide Sequence Design
1 MadSBM reframes peptide design as a rate-based transport problem on an amino-acid edit graph, steering generation through high-likelihood neighborhoods instead of forcing it across chemically implausible intermediates.
2 The model couples a frozen ESM-2 language model to supply biologically informed reference dynamics, then learns only a lightweight 50 M-parameter control field that tilts the CTMC rates toward the data distribution.
3 Training collapses to a simple cross-entropy loss on masked tokens; no expensive forward–backward bridge iterations are required, yet the learned control field provably converges to the minimal-action Schrödinger bridge.
4 Generation runs backward in continuous time with a Poisson jump process; only 32 discrete steps yield peptide sequences whose ESM-2 pseudo-perplexity beats a 150 M-parameter discrete diffusion baseline trained on the same data.
5 Objective-guided sampling is introduced for the first time in a discrete Schrödinger bridge: an external binding classifier reranks 16 candidates per jump, raising predicted affinities above known experimental binders for three of four tested targets.
6 Ablations show that time-gating the ESM prior is critical: removing it increases perplexity, while fully dropping ESM forces the DiT to learn biophysics from scratch, validating the hybrid prior-plus-control design.
7 Theoretical guarantees relate cross-entropy minimization to action minimization, bound control error in KL divergence, and prove convergence of the time-discretized sampler as step size → 0.
💻Code: github.com/pranamchatterjee-…
📜Paper: arxiv.org/abs/2601.22408
#peptidedesign #generativemodeling #schrodingerbridge #proteinlanguageModel #optimaltransport
9
41
2,626
Text2Structure3D: Graph-Based Generative Modeling of Equilibrium Structures with Diffusion Transformers
1. This novel research introduces Text2Structure3D, a novel AI model that generates equilibrium structures from natural language prompts. It leverages a combination of latent diffusion, Variational Graph Auto-Encoders (VGAE), and graph transformers to create structures that adhere to static equilibrium, marking a significant step forward in integrating AI into structural design workflows.
2. The model is designed to support intuitive design exploration by allowing users to specify structural features through text. This approach not only enhances design flexibility but also improves generalization capabilities compared to traditional parametric models, which often require extensive retraining for new applications.
3. A key innovation is the residual force optimization post-processing step, which ensures that the generated structures fully satisfy static equilibrium. This addresses a critical challenge in structural design, where maintaining equilibrium is essential for functional and safe structures.
4. The training dataset consists of a diverse collection of funicular and truss bridge structures paired with detailed text descriptions. This cross-typological dataset enables the model to learn a wide range of structural forms and their corresponding attributes, enhancing its ability to generate accurate and diverse designs.
5. Results demonstrate that Text2Structure3D can generate structures that closely match text-based specifications, even with complex prompts containing multiple attributes. The model shows robustness against variations in text wording, making it highly adaptable for practical design applications.
6. Future work includes expanding the dataset to include more bridge typologies and exploring alternative generative modeling strategies to further enhance the model's scalability and applicability. The authors also suggest potential extensions to other modalities, such as generating structures from images, which could broaden the model's utility in various design contexts.
📜Paper: arxiv.org/abs/2601.12870
#AI #StructuralDesign #GenerativeModeling #DiffusionModels #GraphNeuralNetworks #EquilibriumStructures

5
23
1,441

5 Dec 2025
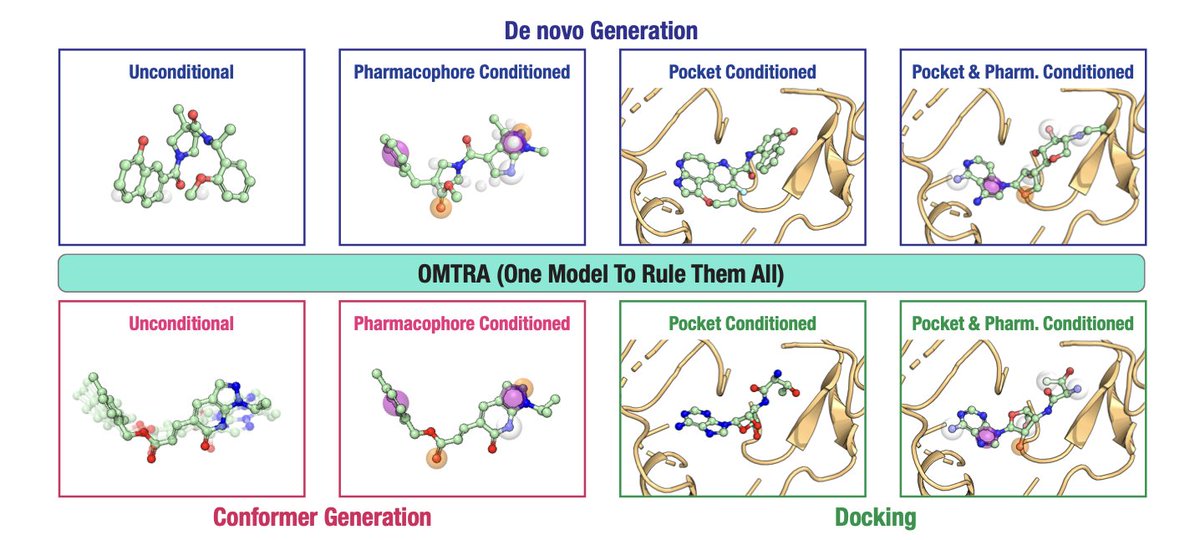
OMTRA: A Multi-Task Generative Model for Structure-Based Drug Design
1. OMTRA introduces a novel multi-task generative model that unifies various tasks in structure-based drug design (SBDD) under one framework. This includes de novo ligand design, docking, and conformer generation, showcasing versatility in handling multiple SBDD-related tasks.
2. The model leverages flow matching, a powerful technique for interpolating between probability distributions, to generate molecular structures. This approach enables the simultaneous modeling of continuous and discrete molecular properties, such as atom positions and types.
3. A key innovation is the ability to condition the model on protein pockets and pharmacophores, allowing for guided design and docking. This feature enhances the accuracy of ligand generation by incorporating prior knowledge of protein-ligand interactions.
4. OMTRA achieves state-of-the-art performance in pocket-conditioned de novo design and docking, outperforming existing models in terms of both chemical plausibility and interaction recovery with proteins.
5. The authors curate a large-scale dataset of 500 million 3D molecular conformers, significantly expanding the chemical diversity available for training. This dataset complements existing protein-ligand data and supports multi-task learning across diverse molecular modalities.
6. Despite the potential benefits of multi-task training, the study finds that its effects are modest and sometimes inconsistent across different tasks. This highlights the ongoing challenge of effectively leveraging transfer learning in molecular generative models.
7. Future work will explore extending OMTRA to generate protein structures, which could support tasks involving flexible or unknown protein conformations. This extension could make OMTRA a more comprehensive tool for realistic drug design scenarios.
💻Code: github.com/gnina/OMTRA
📜Paper: arxiv.org/abs/2512.05080
#DrugDesign #GenerativeModeling #FlowMatching #MultiTaskLearning #StructureBasedDrugDesign #MolecularGeneration
1
4
28
2,193
30 Nov 2025
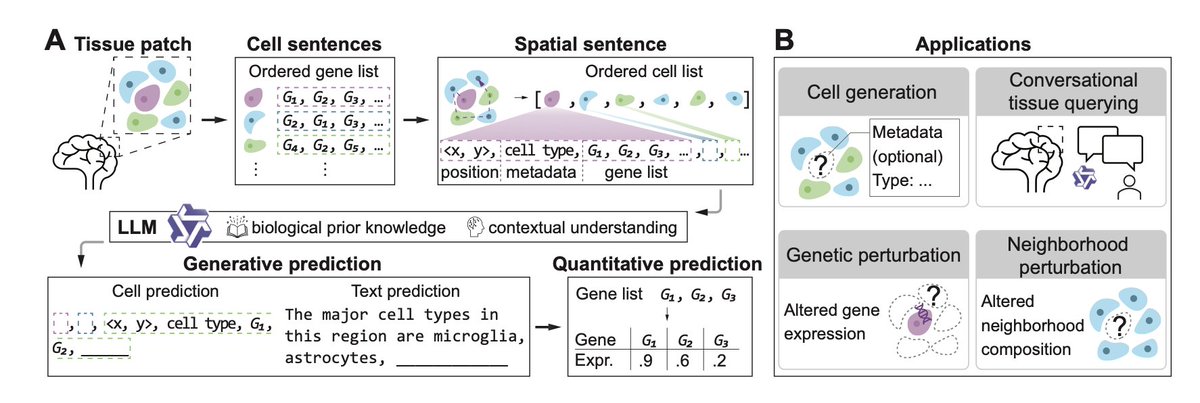
TissueNarrator: Generative Modeling of Spatial Transcriptomics with Large Language Models
1. TissueNarrator introduces a novel approach to spatial transcriptomics by leveraging large language models (LLMs) to simulate cell behavior in situ. This innovative framework transforms tissue sections into "spatial sentences," integrating spatial coordinates and metadata with gene expression data to enable context-aware cell generation.
2. The study demonstrates superior performance in capturing biologically meaningful interactions, such as non-neuronal cell interactions and immune-cancer communication, across multiple spatial transcriptomics technologies like MERFISH, Perturb-FISH, and CosMx SMI.
3. TissueNarrator supports in silico perturbation analyses, allowing researchers to model how cells respond to changes in their neighborhood or genetic modifications. This capability provides a powerful tool for exploring tissue dynamics and disease mechanisms without costly in vivo experiments.
4. A key innovation is the use of explicit numerical tokens for spatial coordinates, bypassing the need for de novo representation learning and activating the LLM's pretrained geometric priors. This design choice enhances the model's ability to interpret spatial information.
5. The framework also enables conversational tissue querying, allowing users to ask natural language questions about tissue organization and receive biologically grounded answers. This feature bridges the gap between data-driven modeling and user-friendly exploration.
6. TissueNarrator's flexible and extensible design supports diverse downstream applications, including cell generation, neighborhood perturbation, genetic perturbation, and interactive Q&A tasks. This versatility makes it a valuable tool for spatial biology research.
7. The study highlights the potential for scaling up model capacity and dataset diversity to enhance TissueNarrator's capabilities further. Larger models could unlock advanced reasoning abilities, while improved context-selection strategies could enable modeling of multi-scale tissue phenomena.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/Steven51516/tissu…
#SpatialTranscriptomics #LargeLanguageModels #GenerativeModeling #ComputationalBiology #TissueNarrator #InSilicoPerturbation #SpatialBiology
2
1,108
18 Nov 2025
BioCG: Constrained Generative Modeling for Biochemical Interaction Prediction
1. BioCG introduces a novel approach to predicting biochemical interactions by reformulating the task as a constrained sequence generation problem. This method leverages a finite catalog of valid target entities, ensuring all outputs correspond to known biochemical options.
2. A key innovation is the Iterative Residual Vector Quantization (I-RVQ) technique, which generates unique discrete sequences for target entities. This allows the model to focus on critical distinctions between valid biochemical options during training and inference.
3. BioCG employs a trie-guided constrained decoding mechanism. This ensures that the model's learning is concentrated on valid biochemical sequences, significantly improving generalization to unseen entities in cold-start scenarios.
4. The framework achieves state-of-the-art performance across multiple biochemical prediction tasks, including Drug-Target Interaction (DTI), Drug-Drug Interaction (DDI), and Enzyme-Reaction Prediction. Notably, BioCG attains an AUC of 89.31% on unseen proteins in the BioSNAP DTI benchmark, a 14.3% improvement over prior methods.
5. An information-weighted training objective enhances learning efficiency by focusing on the most critical decision points. This approach is particularly effective in data-scarce conditions, making BioCG a robust and data-efficient solution for in-silico biochemical interaction prediction.
6. The authors provide open-source code and detailed implementation instructions, facilitating reproducibility and further research in this domain.
📜Paper: openreview.net/pdf/e888694d2…
#Bioinformatics #MachineLearning #DrugDiscovery #GenerativeModeling #BioCG

1
5
1,102
17 Nov 2025
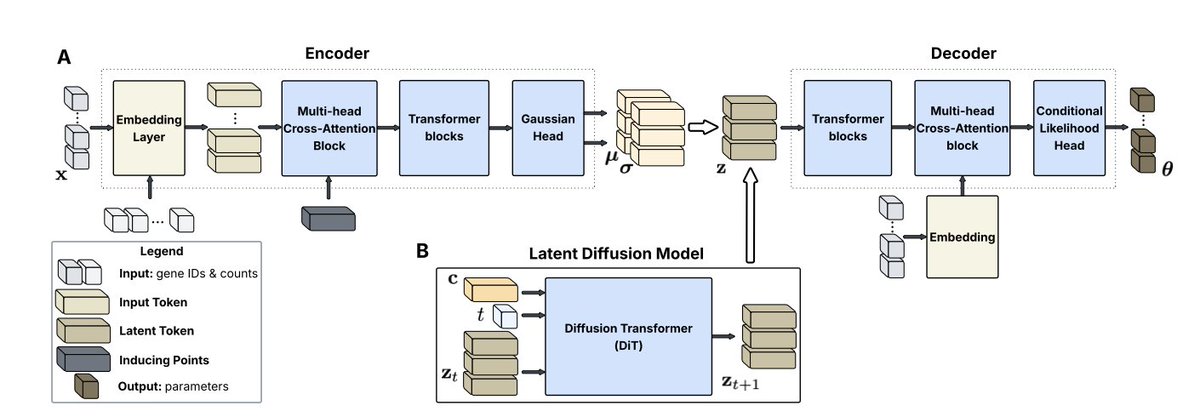
Scalable Single-Cell Gene Expression Generation with Latent Diffusion Models
1. This paper introduces scLDM, a novel latent diffusion model for generating single-cell gene expression profiles. It addresses key challenges in existing models, such as artificial gene orderings and training instabilities, by leveraging a fully transformer-based architecture and a latent diffusion framework.
2. The core innovation lies in the use of a Multi-head Cross-Attention Block (MCAB) that serves dual purposes: permutation-invariant pooling in the encoder and permutation-equivariant unpooling in the decoder. This design respects the exchangeable nature of gene expression data, allowing for high-quality and scalable generation.
3. scLDM replaces the standard Gaussian prior with a latent diffusion model using Diffusion Transformers and linear interpolants. This enables controlled generation through classifier-free guidance, allowing the model to generate cell states conditioned on multiple attributes simultaneously.
4. The model demonstrates superior performance in reconstructing and generating single-cell data across various benchmark datasets. It achieves higher Pearson correlation coefficients and lower reconstruction errors compared to existing methods like scVI and CFGen.
5. In conditional generation tasks, scLDM shows its ability to generate realistic cell states under different perturbations and cell contexts. This is crucial for understanding cellular responses to genetic and chemical perturbations.
6. The embeddings learned by scLDM's VAE also prove useful in downstream classification tasks, such as identifying infected cells in COVID-19 datasets and classifying cell types in Tabula Sapiens 2.0. This highlights the model's potential for biological discovery.
7. The authors provide detailed experiments and ablation studies, demonstrating the model's robustness and the impact of different architectural choices. They also compare different classifier-free guidance strategies, showing that joint conditioning outperforms additive conditioning in multi-attribute settings.
💻Code: github.com/czi-ai/scLDM
📜Paper: arxiv.org/abs/2511.02986
#SingleCellRNASeq #LatentDiffusion #GenerativeModeling #ComputationalBiology #AIinBiology
1
8
1,124
12 Nov 2025
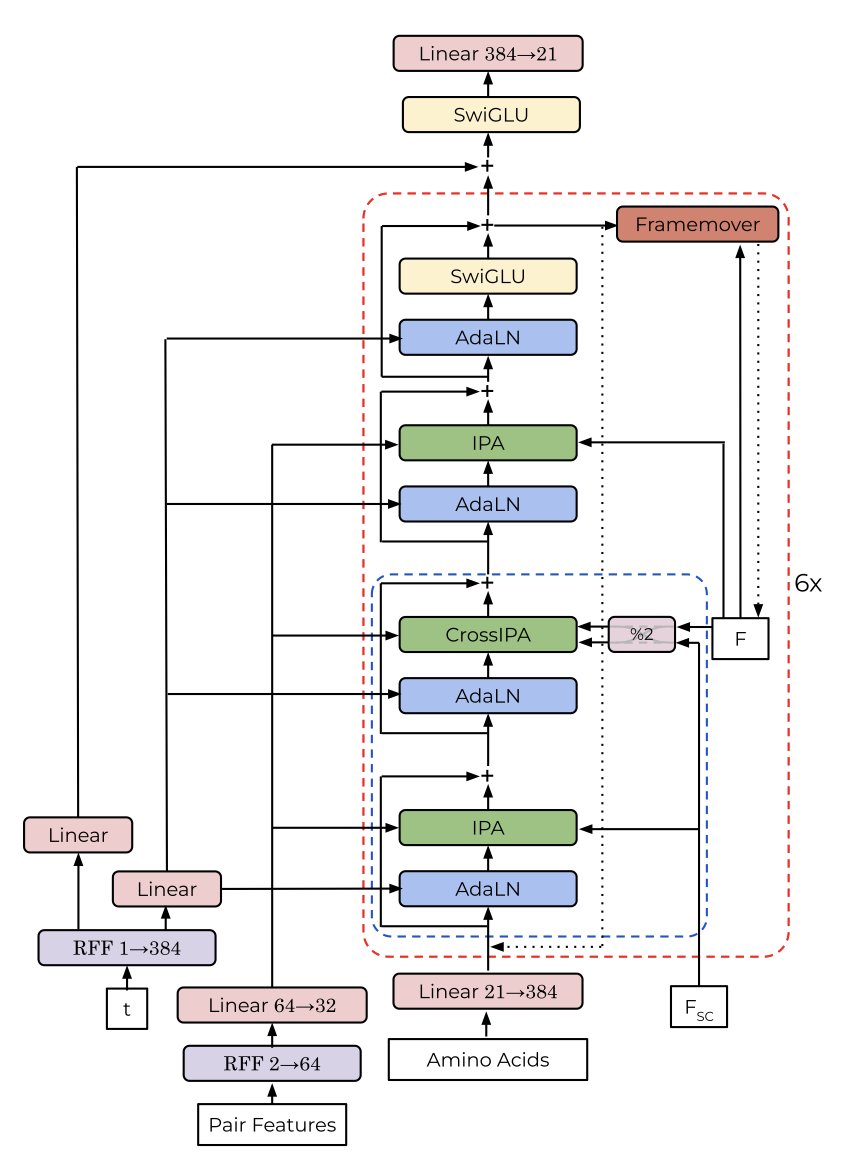
Spontaneous Emergence of Symmetry in a Generative Model of Protein Structure
1. A new study reveals that a transformer-based generative model can spontaneously generate symmetric protein structures without explicit symmetry constraints. This is a significant breakthrough in protein design as it demonstrates the model's ability to learn complex structural patterns implicitly from training data.
2. The model, called ChainStorm, uses flow matching and an SE(3)-equivariant transformer architecture. It was trained on multi-chain protein structures from the Protein Data Bank (PDB) without any symmetry-specific terms in the training objective. The emergence of symmetry was found to be driven by a single attention head in the model, which activates for pairs of residues that match across symmetric chains or repeating motifs within a chain.
3. The researchers discovered that the symmetry in generated structures is primarily determined early in the generative process and is influenced by matching chain lengths. When the lengths of the chains are similar, the model is more likely to generate symmetric structures. This finding suggests that the model can infer symmetry from basic structural information like chain length.
4. The study also demonstrates that suppressing the specific attention head responsible for symmetry prevents the model from generating symmetric structures, confirming its crucial role in symmetry emergence. This highlights the importance of understanding and controlling individual components of generative models to achieve desired design outcomes.
5. ChainStorm's ability to generate symmetric structures without explicit constraints opens new avenues for protein engineering. It suggests that future models could be designed to exploit such emergent behaviors for better control over protein design tasks, including the creation of multimers and self-assembling complexes.
6. The model was implemented in Julia and trained on a dataset of protein structures from the PDB. The training involved a novel approach to sampling and loss scaling, which helped stabilize the learning process and improve the model's performance in generating high-quality protein structures.
📜Paper: biorxiv.org/content/10.1101/…
💻Code: github.com/MurrellGroup/Chai…
#ProteinDesign #GenerativeModeling #Symmetry #TransformerModel #ComputationalBiology
1
3
8
1,124
6 Nov 2025
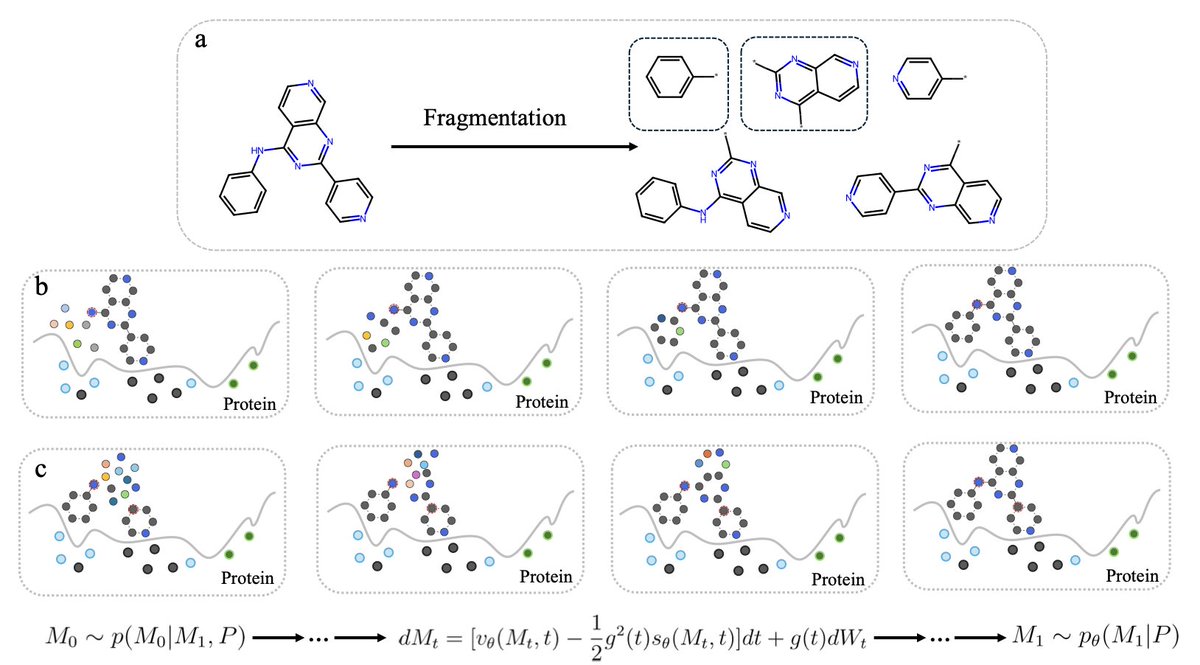
Coupled Fragment-Based Generative Modeling with Stochastic Interpolants
1. This novel study introduces a novel approach to fragment-based drug design using stochastic interpolants, unifying diffusion and flow matching paradigms. The method significantly enhances the efficiency and quality of molecular fragment generation for structure-based drug discovery.
2. The core innovation lies in the explicit fragment-based conditioning of the generative model. Unlike traditional unconditional models, this approach directly trains on molecular substructures, resulting in superior molecular validity and more accurate 3D poses with reduced strain energies.
3. The study demonstrates that flow matching outperforms diffusion models in terms of convergence speed and quality of generated molecules. Flow matching requires fewer computational steps while producing higher-quality 3D molecular structures, making it a more efficient choice for drug design applications.
4. A detailed case study on the PLK3 inhibitor target showcases the model’s ability to generate fragments with competitive binding energies and favorable docking scores compared to existing compounds. This highlights the potential for exploring new chemical space beyond traditional fragment libraries.
5. The choice of fragmentation algorithm is shown to be crucial for model performance. The custom cuttable fragmentation algorithm used in this study generates more diverse fragments, leading to better overall performance in fragment-based drug design tasks.
6. The authors plan to open-source their code and data in an upcoming version of the preprint, facilitating further research and application in the field of computational drug design.
📜Paper: doi.org/10.26434/chemrxiv-20…
#GenerativeModeling #FragmentBasedDrugDesign #StochasticInterpolants #FlowMatching #DrugDiscovery #MolecularGeneration
2
12
1,124
4 Nov 2025
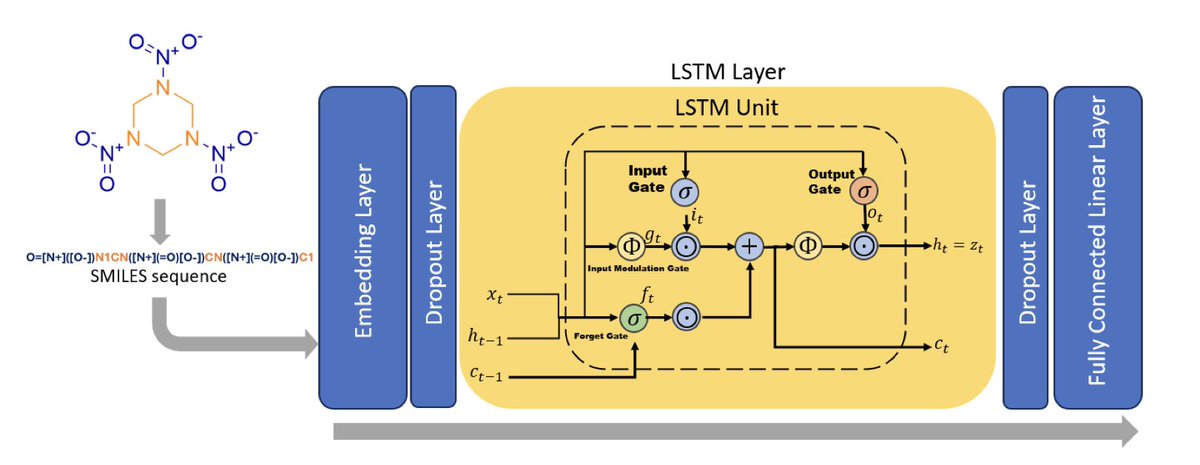
SHA-256 Infused Embedding–Driven Generative Modeling of High-Energy Molecules in Low-Data Regimes
1. This novel study introduces a novel approach to generating high-energy molecules using a combination of LSTM networks for molecular generation and Attentive Graph Neural Networks (GNN) for property predictions. The method is particularly innovative for its ability to work effectively in low-data regimes, a common challenge in the discovery of high-energy materials.
2. The researchers propose a transformative embedding space construction strategy that integrates fixed SHA-256 embeddings with partially trainable representations. This unique approach changes the representational basis itself, reshaping the molecular input space before learning begins, which is a significant departure from conventional regularization techniques.
3. Without pretraining, the generator achieves 67.5% validity and 37.5% novelty, demonstrating the effectiveness of the proposed method even with limited data. The generated library exhibits a mean Tanimoto coefficient of 0.214 relative to the training set, indicating the framework’s ability to generate a diverse chemical space.
4. The study identifies 37 new super explosives with predicted detonation velocities higher than 9 km/s, showcasing the potential of the model to discover novel and high-performing energetic materials. This is a major step forward in the field of high-energy materials discovery.
5. The use of SHA-256-based embeddings provides a deterministic and structured representation that enhances the model’s ability to explore chemically novel yet syntactically valid regions of SMILES space. This embedding strategy is shown to improve generalization and enhance the diversity of generated outputs.
6. The prediction model leverages the AttentiveFP architecture, which outperforms several established approaches in predicting molecular properties. It achieves high R2 values across various properties, demonstrating its robustness and accuracy in multi-target prediction tasks.
7. The study highlights the trade-off between precision and exploration in molecular generation. While models like ULMFiT show stronger alignment with desirable regions, they may limit the discovery of novel molecular scaffolds. The proposed model balances this trade-off effectively.
8. The research underscores the viability of pursuing high-performance material discovery without reliance on pretraining or large-scale chemical corpora, making it accessible for independent research groups and academic laboratories with limited resources.
📜Paper: arxiv.org/abs/2510.25788v1
#HighEnergyMolecules #GenerativeModeling #LowDataRegimes #MachineLearning #MolecularDiscovery
2
5
1,122

31 Oct 2025
🧠 Job Openings: full-time and intern Research Scientist && Engineer (Generative & Multimodal AI),📍 Beijing && Shanghai. Xiaohongshu (RedNote) hi lab.
My Team at Xiaohongshu Hi Lab is Hiring top-tier talents on large-scale text-to-image generative models. We are eager to find experts with strong tracked records on each of these expertise: (1) Data Construction; (2) Model Architecture: Transformer, MoE, etc; (3) Training Methods: diffusion, flow-matching, mean-flow, distillation, consistency, post-training, RLHF, etc; (4) Evaluation System: Benchmark construction, reward modeling, etc; (5) High-performing Infra: Megatron related techniques.
📌 Why you will do?
• Owning a part in training top-tier large generative models that reason, understand, and generate across modalities.
• Publish high-impact papers at top venues (NeurIPS, ICML, ICLR, CVPR, ICCV/ECCV, ACL, TPAMI, etc).
• Joining hi lab at RedNote (Xiaohongshu), an talented and growing lab that aims to explore boundaries of AGI.
BTW, you do not need a PhD title to join us!
NOTE: the opening will close without statement if all positions were taken. Please DM me or send your CV research statement to: pkulwj1994 [at] icloud [dot] com.
#AI #GenerativeModeling #MLResearch #MultimodalAI #BigIdeas #Hiring

1
5
1,078
30 Oct 2025
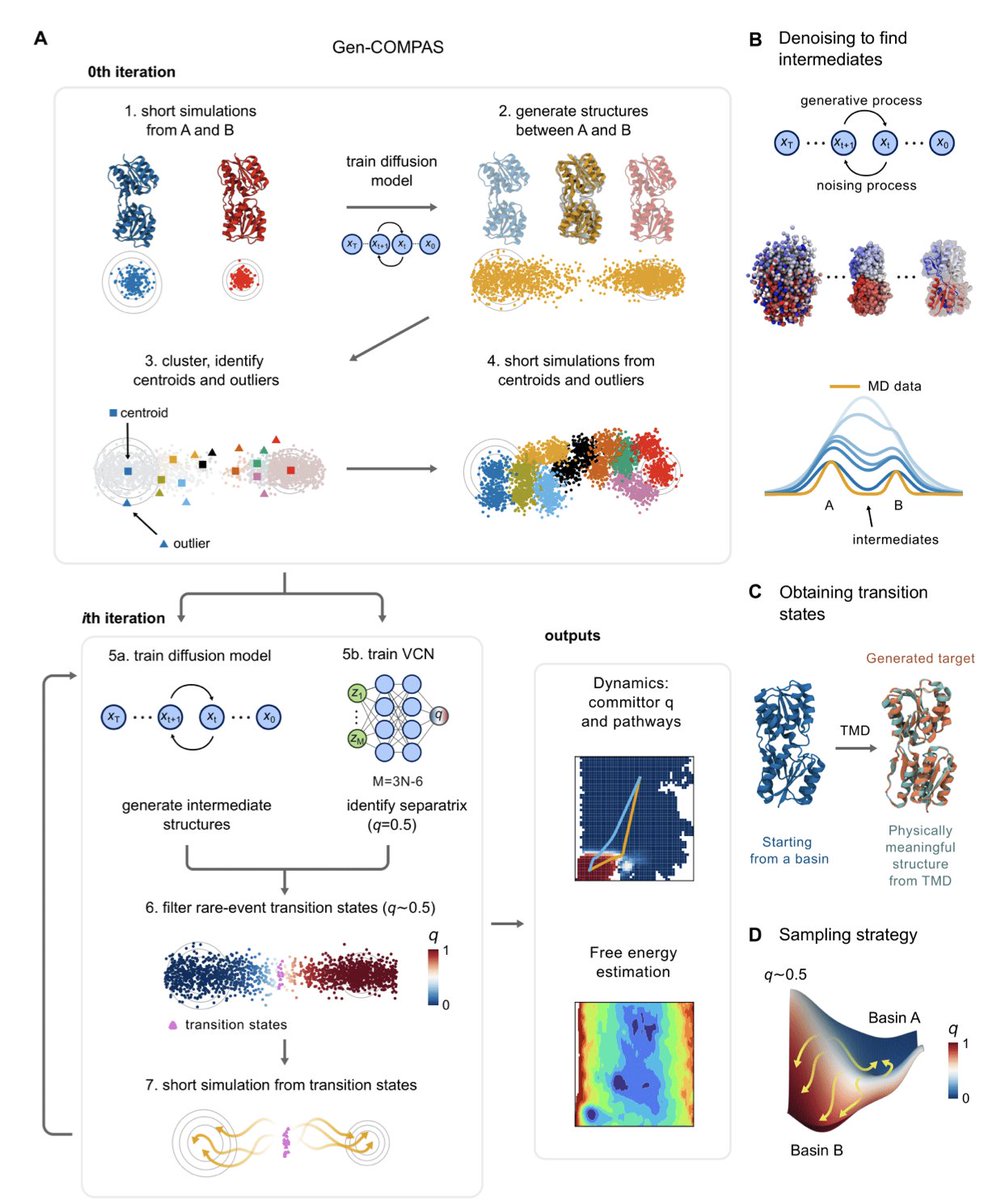
Breaking the Timescale Barrier: Generative Discovery of Conformational Free-Energy Landscapes and Transition Pathways
1. A groundbreaking study introduces Gen-COMPAS, a novel framework that uses generative modeling and committor-guided path sampling to explore rare molecular events. This method reconstructs transition pathways and free-energy landscapes without predefined variables, significantly reducing computational costs compared to traditional molecular dynamics simulations.
2. Gen-COMPAS leverages a diffusion model to generate physically realistic intermediate conformations and uses committor-based filtering to identify transition states. This iterative approach allows for rapid convergence of transition-path ensembles within nanoseconds, a dramatic improvement over conventional methods that require orders of magnitude more sampling.
3. The framework is demonstrated on systems ranging from a miniprotein to a ribose-binding protein and a mitochondrial carrier. It efficiently retrieves committor maps, transition states, and free-energy landscapes, providing deep mechanistic insights and practical applications in drug discovery and protein design.
4. A key innovation of Gen-COMPAS is its ability to model protein-ligand complexes and other heterogeneous biomolecular assemblies without adaptation. This capability, combined with its GPU optimization, makes it scalable to large biomolecular systems and applicable to a wide range of biological processes.
5. The study shows that Gen-COMPAS can reduce sampling times by hundreds of times, making it an exceptionally efficient tool for investigating rare events in complex systems. It also provides a unified approach to studying both thermodynamics and kinetics, offering a comprehensive view of molecular transitions.
6. The framework's efficiency and accuracy are demonstrated through applications to fast-folding proteins like Trp-cage, binding-upon-folding processes in ribose-binding proteins, and the conformational transitions of the mitochondrial ADP/ATP carrier. These examples highlight Gen-COMPAS's potential to transform our understanding of molecular dynamics.
📜Paper: arxiv.org/abs/2510.24979
#MolecularDynamics #GenerativeModeling #ComputationalBiology #RareEvents #FreeEnergyLandscapes #TransitionPathways
1
6
31
2,395
22 Oct 2025
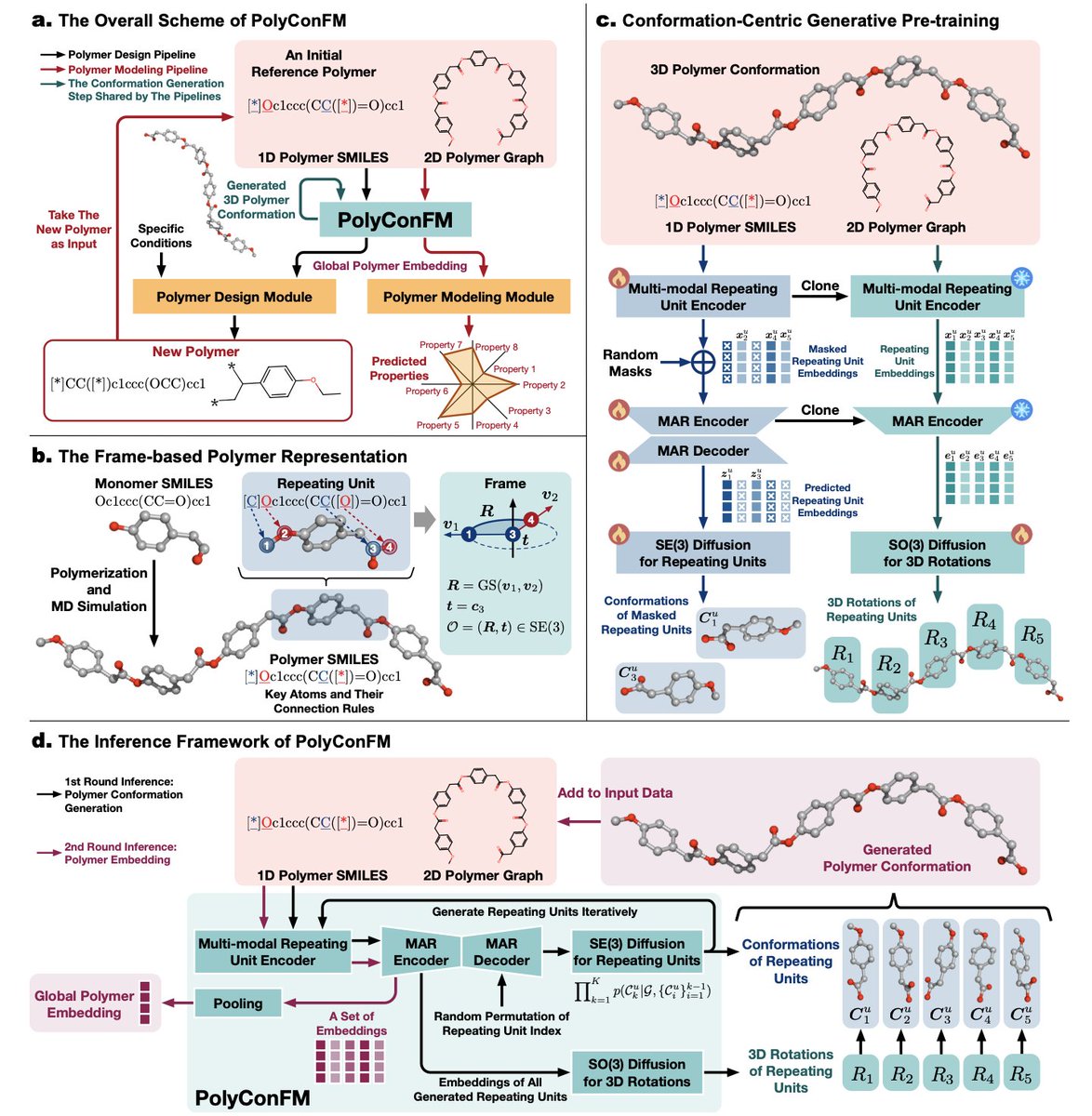
Unifying Polymer Modeling and Design via a Conformation-Centric Generative Foundation Model
1. The article introduces PolyConFM, a novel polymer foundation model that integrates polymer modeling and design through a novel conformation-centric generative pretraining approach. This model addresses the limitations of existing methods by capturing global structural information inherent in polymer conformations, which is crucial for accurate polymer modeling and design.
2. PolyConFM innovatively decomposes polymer conformations into sequences of local conformations (repeating units) and uses masked autoregressive modeling to reconstruct these local conformations. It further generates orientation transformations to recover the complete polymer conformation, effectively capturing complex dependencies among repeating units.
3. A significant contribution of this work is the construction of a high-quality dataset of over 50,000 polymers with conformations obtained through molecular dynamics simulations. This dataset not only enables conformation-centric pretraining but also provides a valuable resource for future research in polymer science.
4. Experiments demonstrate that PolyConFM significantly outperforms state-of-the-art methods in polymer conformation generation, property prediction, and design tasks. It achieves state-of-the-art performance by providing accurate global structural information and effectively supporting diverse downstream tasks, thereby bridging the gap between polymer structure, property, and design.
5. The conformation-centric generative pretraining approach of PolyConFM is particularly noteworthy. It leverages the inherent 3D structures of polymers to generate informative representations that are essential for both property prediction and design. This method sets a new standard for polymer foundation models by incorporating global structural features that are often overlooked in existing methods.
📜Paper: arxiv.org/abs/2510.16023v1
#PolymerScience #DeepLearning #GenerativeModeling #Conformation #PolymerDesign #FoundationModel
1
8
871
15 Oct 2025
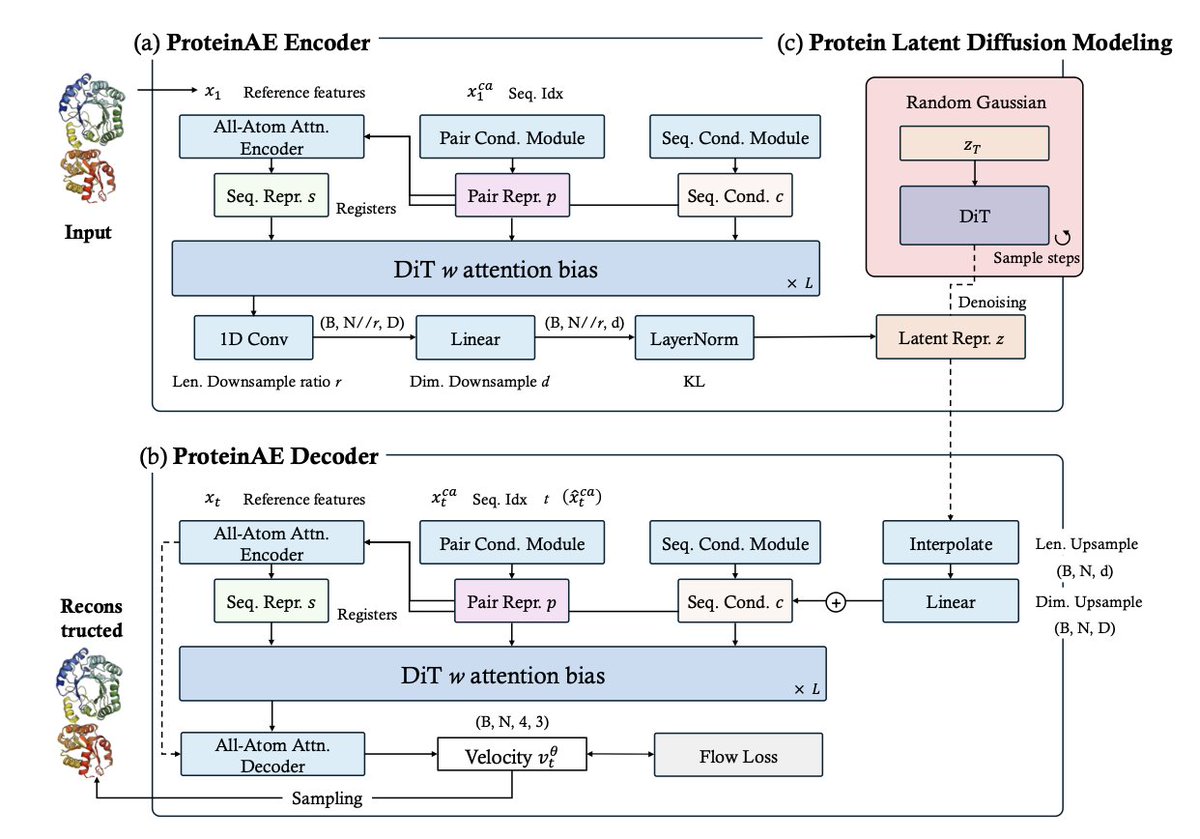
Proteinae: Protein Diffusion Autoencoders for Structure Encoding
1. Proteinae introduces a novel protein diffusion autoencoder that maps protein backbone structures into a continuous, compact latent space using a non-equivariant Diffusion Transformer architecture. This approach avoids the complexities of the SE(3) manifold and discrete representations inherent in existing methods.
2. The model employs a simple flow matching objective for training, which maximizes the ELBO of the likelihood of the input protein structures. This single training objective simplifies the optimization pipeline and leads to state-of-the-art reconstruction quality.
3. Proteinae achieves high-fidelity protein structure reconstruction, outperforming existing autoencoders on the CASP14 and CASP15 benchmarks. The learned latent space serves as a powerful foundation for downstream tasks such as protein latent diffusion modeling (PLDM) and physicochemical property prediction.
4. The resulting PLDM is competitive with leading structure-based generative models and significantly outperforms prior latent-based methods in both sample quality and efficiency. This demonstrates the potential of Proteinae for efficient and high-quality protein structure generation.
5. The study also includes an ablation study on key architectural components, such as the impact of protein length and dimension bottlenecks, model scalability, and the use of registers for latent compression. These analyses provide valuable insights into the design and optimization of protein autoencoders.
6. Despite its capabilities, Proteinae currently has limitations, including its focus on protein monomers and challenges related to sequence length handling in PLDM. Future work will aim to address these limitations and explore extensions to the model.
📜Paper: arxiv.org/abs/2510.10634v1
#Proteinae #ProteinStructure #DiffusionAutoencoder #GenerativeModeling #ComputationalBiology
1
2
34
2,180
10 Oct 2025
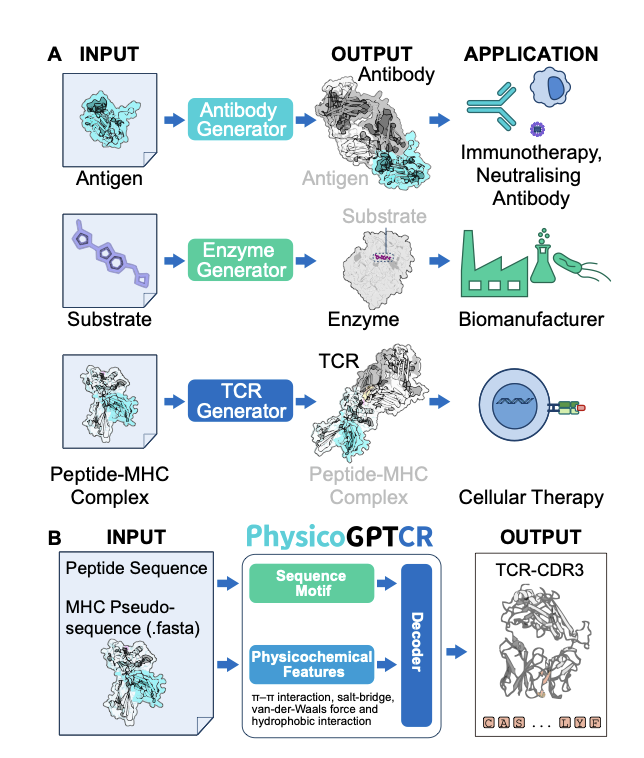
Physicochemically Informed Dual - Conditioned Generative Model of T - Cell Receptor Variable Regions for Cellular Therapy
1. A novel study introduces PhysicoGPTCR, a novel generative model for designing T - cell receptor (TCR) variable regions. This model is uniquely dual - conditioned on both peptide and HLA context, embedding residue - level physicochemical descriptors to ensure biophysical plausibility and improve the generation of functional TCR candidates.
2. The model leverages a Transformer - based architecture, fusing token identity, positional index, and physicochemical features through a learnable gating mechanism. This approach allows the network to reason about long - range chemical bonds during sequence generation, addressing a critical gap in existing models.
3. PhysicoGPTCR outperforms competitive baselines such as ANN, GPTCR, LSTM, and VAE across multiple metrics. It achieves lower edit - distance, higher sequence similarity, and longer common subsequence lengths, demonstrating superior performance in generating TCR sequences that closely match experimentally verified receptors.
4. The study includes a robust evaluation through in - silico docking and structural modeling, revealing a higher proportion of binding - competent clones compared to the strongest baseline. This validates the model's ability to produce functional TCR candidates with high specificity for neoantigens.
5. Ablation studies confirm the importance of incorporating physicochemical embeddings, showing that removing this component leads to worse performance in all metrics. This highlights the value of explicit context conditioning and physicochemical awareness in the model.
6. The model generalizes well across diverse immunological contexts, including twelve HLA class I alleles and eight canonical viral and tumor - associated epitopes. This suggests broad applicability in generating TCRs for various clinical scenarios.
7. The study also demonstrates the model's potential for rapid, personalized TCR design, reducing the discovery timeline from months to minutes. This could significantly accelerate the development of targeted cancer immunotherapies and vaccines.
📜Paper: arxiv.org/abs/2510.05747v1
#ComputationalBiology #TCRDesign #GenerativeModeling #Immunotherapy #AIinMedicine
1
11
1,253
25 Sep 2025
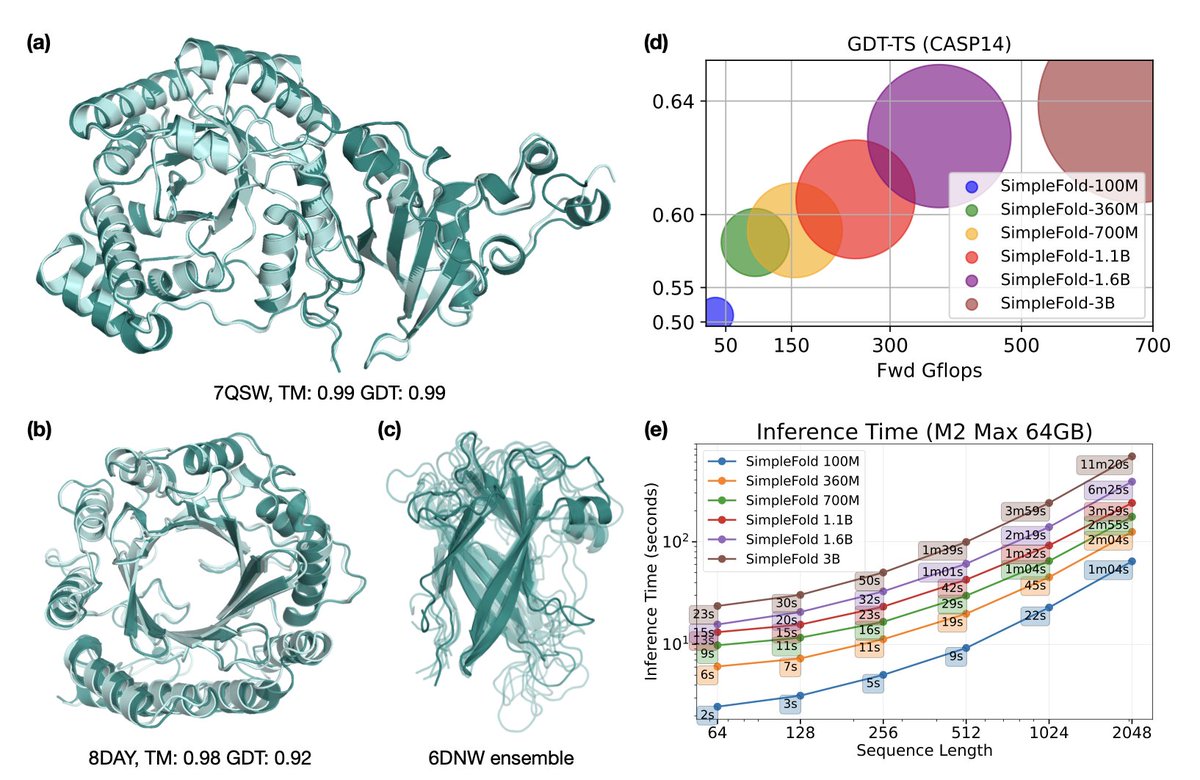
SimpleFold: Folding Proteins is Simpler than You Think
1. A novel protein folding model called SimpleFold has been introduced by researchers from Apple. This model achieves remarkable performance using a general-purpose transformer architecture, challenging the necessity of complex domain-specific designs in protein folding.
2. SimpleFold is the first flow-matching based protein folding model that relies solely on standard transformer blocks. It eliminates the need for computationally expensive modules like triangular updates, explicit pair representations, and multiple training objectives, simplifying the architecture significantly.
3. The model is trained with a generative flow-matching objective and an additional structural term. It scales to 3B parameters and is trained on approximately 9M distilled protein structures along with experimental PDB data, demonstrating strong performance on standard folding benchmarks.
4. SimpleFold shows competitive performance compared to state-of-the-art baselines and excels in ensemble prediction, which is typically challenging for models trained with deterministic reconstruction objectives. This highlights its potential for generating diverse and accurate protein structures.
5. Due to its general-purpose architecture, SimpleFold is highly efficient in deployment and inference on consumer-level hardware. This makes it accessible for a wide range of applications and researchers without requiring specialized computational resources.
6. The authors release a family of models ranging from an efficient 100M model to a large 3B model, providing flexibility in performance and efficiency trade-offs. This allows researchers to choose the best model size based on their specific needs and computational constraints.
7. SimpleFold challenges the reliance on complex domain-specific architectures in protein folding, opening up a new design space for future progress. It demonstrates that strong folding performance can be achieved without explicit pairwise representations or triangle updates, reducing architectural complexity.
8. The model also includes a confidence module that predicts per-residue LDDT values as a confidence score, providing insights into the quality of generated structures. This can be particularly useful for understanding the reliability of predictions in different regions of the protein.
9. SimpleFold is trained on a mix of PDB, SwissProt, and AFESM data, leveraging the vast amounts of available computationally predicted structures. This large-scale training approach contributes to its robust performance and ability to generalize across diverse protein structures.
10. The results on CAMEO22 and CASP14 benchmarks show that SimpleFold achieves competitive performance with state-of-the-art models, demonstrating its effectiveness in predicting protein structures. It also shows strong performance in ensemble generation tasks, which is crucial for applications requiring modeling of protein flexibility.
11. The study highlights the potential of scaling up general-purpose transformer architectures for protein folding. The positive scaling behavior of SimpleFold suggests that larger models trained on more data can lead to even more powerful and accurate protein structure prediction models in the future.
12. SimpleFold represents a disruptive approach to protein folding, relying on scaling up general-purpose architecture blocks to learn the symmetries of the underlying data generation process directly from training data. This approach could pave the way for more efficient and powerful protein generative models.
💻Code: github.com/apple/ml-simplefo…
📜Paper: arxiv.org/abs/2509.18480v1
#ProteinFolding #SimpleFold #Transformer #GenerativeModeling #ComputationalBiology #AI #MachineLearning
2
29
137
28,281
25 Sep 2025
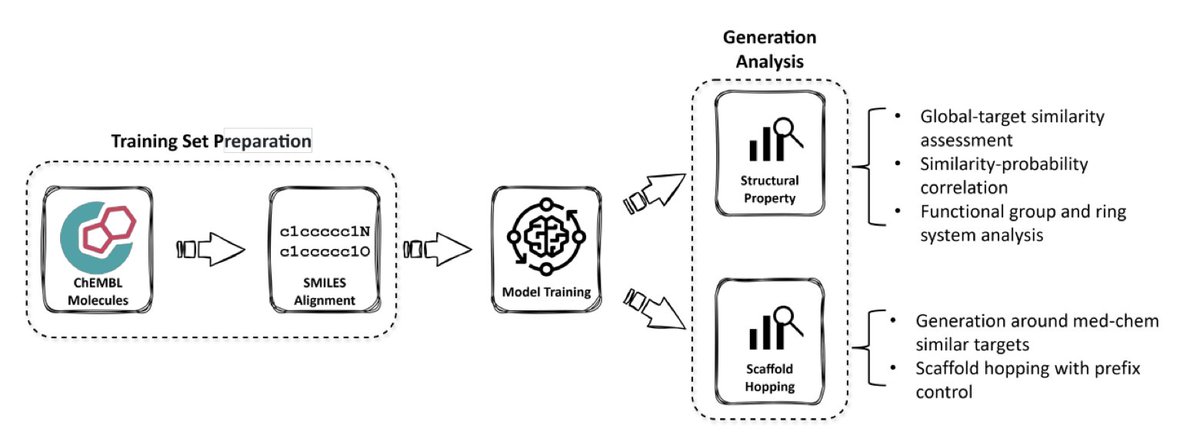
ANNalog — Generation of MedChem-similar Molecules
1. ANNalog is a transformer-based sequence-to-sequence generative model that can produce both structurally similar analogues and perform scaffold hopping, which is crucial for medicinal chemistry. This model is trained on pairs of molecules from the same bioactivity assay in ChEMBL33, capturing the essence of medicinal chemistry analogues.
2. The innovation of ANNalog lies in its training approach. Unlike previous models that rely on computational structural similarity measures, ANNalog uses a medicinal chemistry similarity definition. It considers molecules tested within the same assay as analogues, making it more aligned with practical medicinal chemistry practices.
3. A key feature of ANNalog is its ability to perform scaffold hopping. This was validated using manually curated molecule pairs and further confirmed through a case study involving orexin-2 receptor antagonists from patent literature. When constrained with prefix control, ANNalog successfully rediscovered a significant proportion of known scaffolds.
4. The model incorporates SMILES alignment using Levenshtein distance during preprocessing, which enhances its ability to learn structural transformations. Data augmentation with multiple SMILES representations further improves its robustness and generalisation across different input formats.
5. In terms of performance, ANNalog demonstrates a high degree of structural similarity to input ligands and generates molecules that are relevant to the global targets within assays. It also shows a strong capability for scaffold hopping, outperforming the Mol2Mol model in generating structurally diverse yet chemically relevant analogues.
6. The study highlights that ANNalog can be guided to retain key structural motifs while exploring scaffold variations through prefix control. This makes it a powerful tool for molecular optimisation tasks in AI-driven drug discovery, balancing the generation of similar analogues and scaffold hopping effectively.
📜Paper: doi.org/10.26434/chemrxiv-20…
#GenerativeModeling #MolecularGeneration #ScaffoldHopping #DrugDiscovery #AIinMedChem
1
7
1,282
24 Sep 2025
Guided Sequence-Structure Generative Modeling for Iterative Antibody Optimization
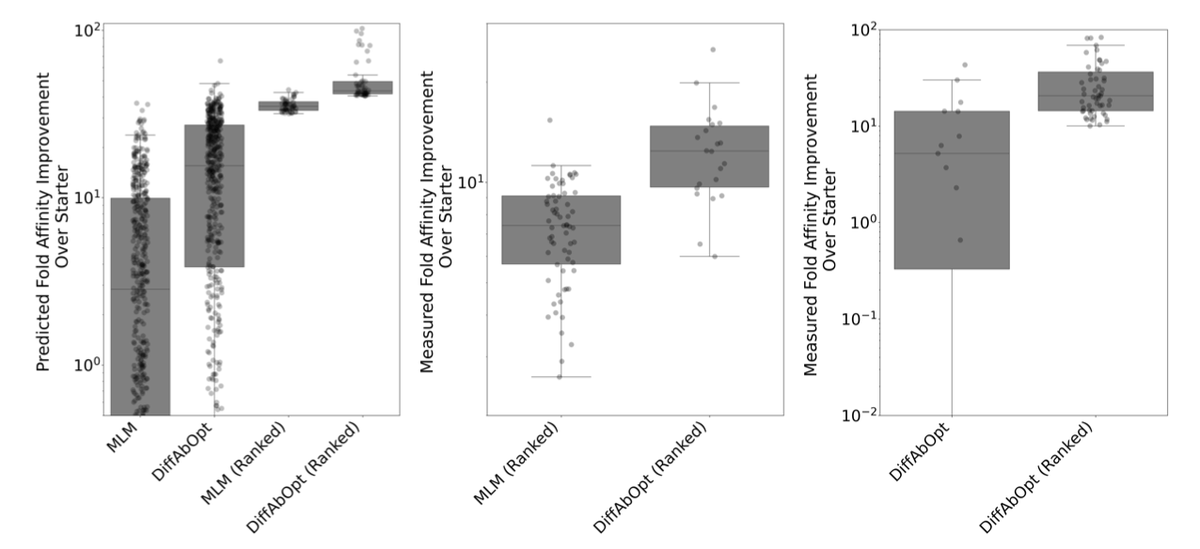
1. This paper proposes a novel approach for iterative antibody optimization that integrates sequence and structure information, addressing the challenge of lacking structural data for evolving lead molecules during optimization.
2. The authors build on the DiffAb model, training a sequence-structure diffusion generative model that operates on antibody-antigen complexes. They outline a pipeline to use predicted Ab-Ag complexes for lead candidates in each design round.
3. A key innovation is the guided sampling method, which biases the generation of antibody sequences toward desirable properties by incorporating oracle models trained on experimental data from iterative design.
4. In silico evaluations show that their method produces distributions of sequences enriched for high binding affinity and low polyreactivity. In vitro experiments further demonstrate the success of their approach, with candidates showing strong binding affinities at multiple stages of optimization.
5. This work highlights the potential of leveraging both sequence and structure information in iterative antibody design, and the effectiveness of guided sampling in generating optimized antibody candidates.
📜Paper: arxiv.org/abs/2509.16357
#AntibodyOptimization #GenerativeModeling #IterativeDesign #ComputationalBiology
1
4
1,149
4 Sep 2025
Morphology-Specific Peptide Discovery via Masked Conditional Generative Modeling
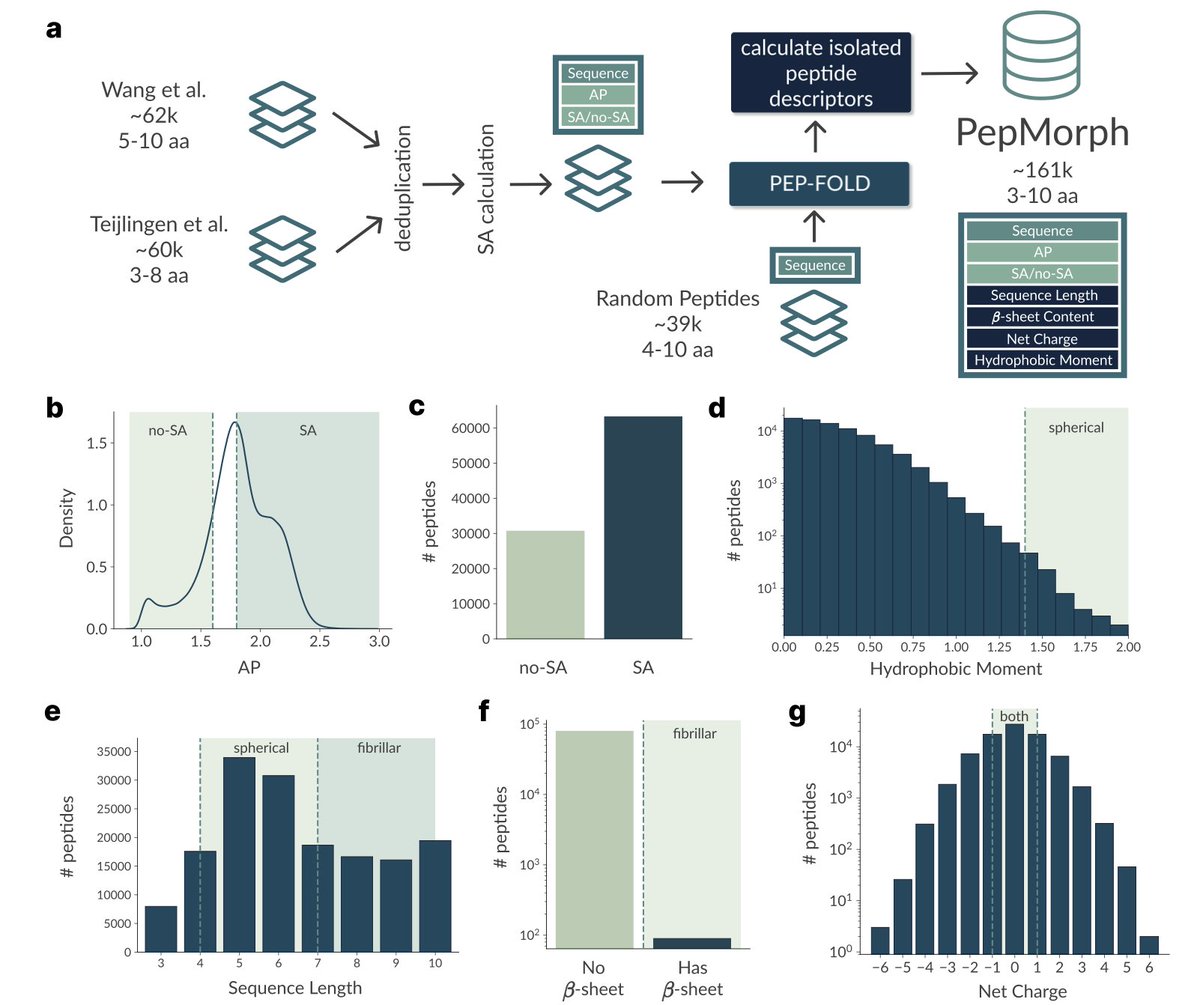
1. A novel study introduces PepMorph, an end-to-end pipeline for discovering peptides that self-assemble into specific morphologies like fibers or spheres. This is a significant step forward in designing biocompatible materials with tailored properties for biomedical and energy applications.
2. PepMorph leverages a Transformer-based Conditional Variational Autoencoder with a masking mechanism. This allows the model to generate novel peptides under arbitrary conditioning, making it highly flexible for targeting different morphologies based on geometric and physicochemical descriptors.
3. The study compiles a new dataset by integrating existing aggregation propensity datasets and extracting peptide descriptors that act as proxies for aggregate morphology. This dataset is crucial for training the model and ensuring it can generate peptides with desired properties.
4. PepMorph demonstrates high novelty and diversity in generated sequences, with less than 10% similarity among generated peptides. The model achieves 83% accuracy in intended morphology generation, validated through coarse-grained molecular dynamics simulations.
5. The framework is highly versatile and can be extended to longer peptides and more complex morphologies. It also allows for the integration of additional descriptors, making it a powerful tool for future peptide design and discovery efforts.
6. The study highlights the importance of the masking mechanism in handling partial conditioning, which is essential for generating peptides that meet specific design criteria without collapsing to a few templates.
7. The authors provide the curated PepMorph dataset and simulation trajectories publicly, along with the trained model weights and code, facilitating further research and development in this exciting field.
💻Code: github.com/tummfm/pepmorph
📜Paper: arxiv.org/abs/2509.02060v1
#PeptideDiscovery #MolecularDesign #GenerativeModeling #BiocompatibleMaterials #MolecularDynamics #TransformerModel #MachineLearning #ComputationalBiology
1
2
11
1,511
3 Sep 2025
MolSculptor: An Adaptive Diffusion-Evolution Framework Enabling Generative Drug Design for Multi-Target Affinity and Selectivity
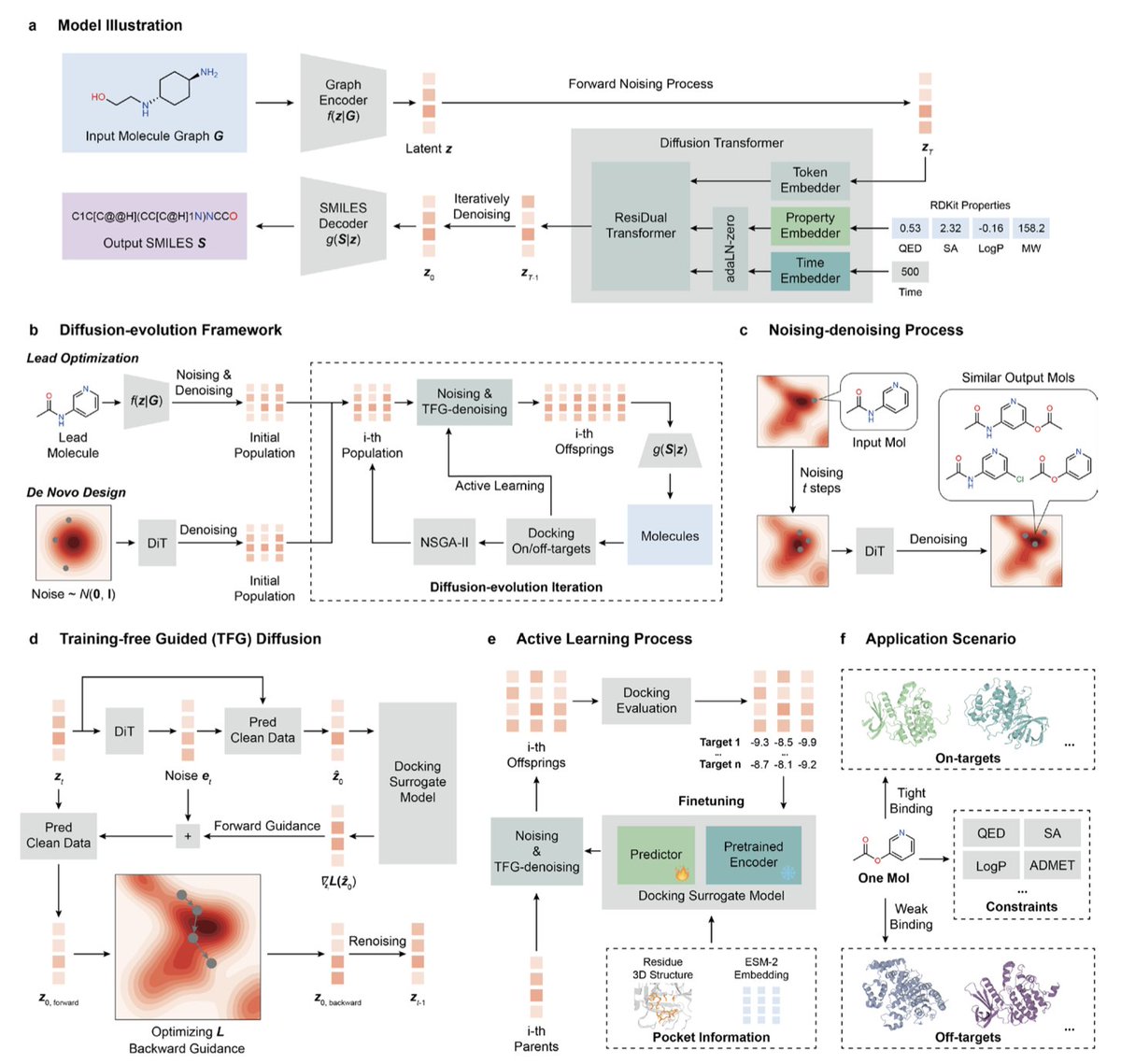
1. MolSculptor introduces a novel adaptive diffusion-evolution framework designed to generate inhibitors for any combination of on-targets and off-targets, bypassing the need for target-specific training data or prior expert knowledge. This innovation addresses a significant challenge in multi-target drug design, making it a powerful tool for complex therapeutic problems.
2. The framework integrates a 3D-aware surrogate model that enables flexible guidance for any set of specified on-targets and off-targets. It employs an active learning protocol to adaptively refine this guidance, ensuring high performance even in data-scarce scenarios. This adaptability is crucial for navigating the vast chemical space efficiently.
3. MolSculptor unifies both de novo design and lead optimization, providing a versatile workflow applicable to different stages of drug discovery. It allows for direct conditioning on key drug-like properties, such as QED, SA, LogP, and MW, to generate molecules that adhere to desired property profiles.
4. Demonstrated on a series of challenging multi-target and selective inhibitor design tasks, MolSculptor significantly outperforms state-of-the-art methods in generating high-quality candidates that satisfy all complex constraints. Many of the generated molecules exhibit predicted affinity profiles superior to those of experimentally validated references.
5. The noising-denoising process in MolSculptor serves as a powerful molecular editing operator, offering fine-grained control over the generative process. The diffusion timestep provides stochastic control over the degree of structural modification, while property conditioning exerts more deterministic control over the type of molecules being generated.
6. In dual-target lead optimization tasks, MolSculptor consistently discovers molecules with significantly better docking scores at every stage of the evolution while satisfying all predefined constraints. This performance advantage is attributed to the training-free guidance (TFG) mechanism, which steers the generation process towards molecules with predicted improvements in desired properties.
7. For selective inhibitor design, MolSculptor successfully navigates the complex landscape of distinguishing between highly similar protein pockets. In both lead optimization and de novo design modes, it generates molecules with clear separation in docking scores between on-targets and off-targets, demonstrating its versatility and efficiency.
8. The modular design of MolSculptor allows for easy integration of different affinity oracles and advanced drug-target affinity measures. Future work will focus on incorporating more sophisticated physical and biological constraints, such as explicit 3D molecular representations and ADMET properties, to further enhance its capabilities.
📜Paper: doi.org/10.26434/chemrxiv-20…
💻Code: github.com/egg5154/MolSculpt…
#MolSculptor #DrugDesign #MultiTarget #SelectiveInhibitors #DiffusionModel #ActiveLearning #ComputationalChemistry #GenerativeModeling
2
26
2,018