Interpretable enzyme function prediction via sparse autoencoder features of ESMC across the microbial protein universe
1. The paper proposes a “features-first” route to enzyme function prediction: instead of training a new deep model, it uses ESMC-6B’s sparse autoencoder (SAE) features as an interpretable semantic signature to predict EC numbers.
2. Key enabler: the ESMC SAE expands layer-60 hidden states into a 16,384-feature codebook with Top-K=64 sparsity, and each feature has an independently interpretable biological concept label (generated/annotated via multi-agent GPT-5), enabling mechanistic explanations alongside predictions.
3. Benchmark setup: 4,868 reviewed microbial SwissProt enzymes (Bacteria Archaea), balanced across 7 EC1 superclasses and spanning 161 EC3 subclasses; proteins are 80–700 aa and have ≥3 EC levels. Protein representations are mean-pooled SAE activations, evaluated with simple linear probes.
4. Main EC3 result (161-way classification, 80/20 split): SAE binary features (just the top-64 active concepts per protein) reach 78.9% top-1 and 88.5% top-5 accuracy, outperforming a 3-mer logistic regression baseline (57.3% top-1) and approaching BLASTp (80.5% top-1).
5. Using richer SAE information improves further: the combined binary activation weights representation reaches 85.6% top-1 and 90.5% top-5, exceeding BLASTp top-1 by 6.4% in this benchmark.
6. Practical advantage over homology transfer: BLASTp returns no hit for 12.6% of test proteins, while SAE features yield predictions for 100% of queries; for the BLASTp no-hit subset, SAE binary still achieves 62.1% top-5 accuracy.
7. “Dark-matter regime” analysis: test proteins are binned by maximum 3-mer Jaccard similarity to training data; in the lowest-similarity bin (<0.20, containing 61% of the test set), SAE top-5 accuracy is 0.656 vs 0.438 for the 3-mer baseline, showing robustness when sequence similarity is minimal.
8. Generalization to unseen enzyme classes: leave-one-EC3-class-out evaluation (60 populous EC3 subclasses held out) trains only an EC1 classifier and tests whether it recovers the correct EC1 superclass for a completely unseen EC3 class; SAE binary achieves 47.7% EC1 recovery (3.3× random; vs 26.6% for sequence features).
9. Interpretability check: the most discriminative SAE features align with known enzymology—e.g., hydrolases linked to α/β-hydrolase catalytic triad/nucleophilic elbow geometry; oxidoreductases to Rossmann NAD(P)H-binding motifs; transferases to P-loop/Walker A phosphate-binding patterns; translocases to multi-helix transmembrane bundle concepts—supporting mechanistic plausibility of the learned “concept” features.
10. Atlas-scale survey: scanning 7.7M ESM Atlas cluster representatives, the authors identify 169,859 “dark enzyme-like” candidates (uncharacterized clusters with enzyme-suggestive Pfam keywords) spanning major microbial phyla, positioning SAE-based signatures as a scalable prioritization tool for experimental enzyme discovery.
💻Code: github.com/YueHuLab/esmc-sae…
📜Paper: arxiv.org/abs/2606.12209
#ProteinLanguageModels #EnzymeDiscovery #ECNumber #Interpretability #SparseAutoencoders #Microbiome #Metagenomics #ComputationalBiology #ESMC #ESMAtlas

5
34
2,254

Jun 5
Check out this press release by LLNL: computing.llnl.gov/about/new…
Please stop by if you’re attending CVPR, would love to discuss interpretability, sparse autoencoders, and concept bottleneck models!
#CVPR2026 #Interpretability #ExplainableAI #SparseAutoencoders #ConceptBottlenecks
1
4
209
SMolLM: Small language models learn small molecular grammar
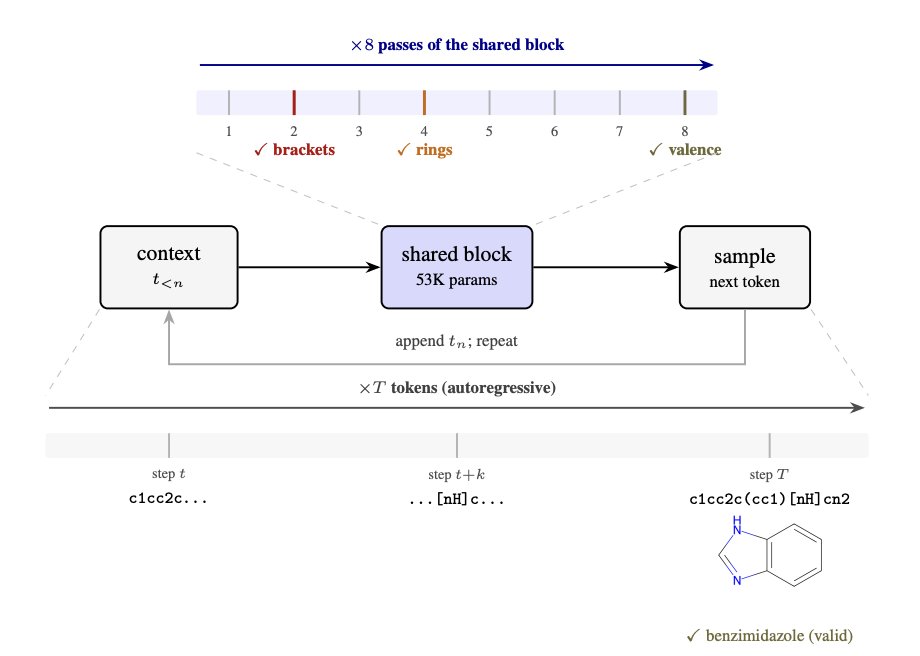
1 SMolLM trains a 53K-parameter weight-shared transformer (one Modern GPT block reused for multiple passes) to generate SMILES with 95.3% RDKit validity on ZINC-250K, beating an identically trained unshared GPT with 10× more parameters (GPT-527K: 87.6% validity).
2 The key idea is trading parameters for iterative computation: for each next-token prediction, the same block is applied repeatedly (virtual depth). Validity climbs sharply with more passes and plateaus around 8 passes (e.g., WS-206K: 33% at D=1 → 90.5% at D=2 → 97.1% at D=4 → 98.6% at D=8).
3 Weight sharing dominates the sub-megaparameter parameter–validity Pareto frontier: across <1M parameters, WS models consistently achieve higher validity than unshared GPT baselines at similar or larger sizes (e.g., WS-53K reaches 95.3%; much larger unshared GPTs are needed to approach that level).
4 Mechanistic result: the same shared block resolves SMILES constraints in a consistent, staged order across passes—brackets first, ring closures second, valence last. This is quantified using exact symbolic checks for each constraint, enabling per-pass “grammar progress” measurement.
5 Output-level error classification shows the stage ordering directly: bracket errors collapse by pass 2 (e.g., 23.0% → 1.3% from D=1 to D=2), ring errors drop next (notably from pass 2 to pass 4), and valence violations are the last major residual class (improving mainly from pass 4 to pass 8).
6 Internal representations mirror the same hierarchy. Linear probes decode bracket depth earlier than ring state (WS-206K: bracket-depth probe peaks at pass 2; ring-state probe peaks at pass 5). Sparse autoencoders (SAEs) recover early token-level detectors (bracket / ring-digit), followed by later compositional state features (bracket depth, ring state), and then more atom-level identity features.
7 Causal localization: a single attention head is responsible for the earliest bracket-matching step. Ablating that head at pass 1 selectively increases bracket errors (WS-206K: 22 percentage points bracket errors; validity −21 pp) while leaving ring/valence errors near baseline—supporting a specific circuit rather than general sensitivity.
8 Capacity mainly changes how many passes are needed, not the ordering. The smaller WS-53K reaches similar milestones ~1–2 passes later and relies on the bracket head across more early passes, suggesting iterative refinement compensates for limited per-pass capacity.
9 Robustness checks: benefits persist across decoding settings (temperature/top-k/top-p) and are not explained by extra training of unshared baselines. Interestingly, standard post-training methods (offline distillation, DPO) reduce validity, implying that preserving per-pass iterative computation may require different objectives than typical output-only alignment.
💻Code: github.com/akhljndl/smollm
📜Paper: arxiv.org/abs/2605.06322
#ComputationalBiology #Cheminformatics #MolecularGeneration #SMILES #Transformers #MechanisticInterpretability #WeightSharing #SparseAutoencoders #DrugDiscovery #MachineLearning
4
18
1,712

22 Dec 2025
Gemma Scope 2 is here: an open “microscope” for Gemma 3, built for real AI safety work.
Released on December 19, 2025, Gemma Scope 2 is Google DeepMind’s new open interpretability suite for the entire Gemma 3 family. This post breaks down what it is, what’s actually included, why the scale matters, and how beginners and experts can use it to investigate tricky model behaviors like jailbreaks and hallucinations.
@GoogleDeepMind
🔗Tap below to dive deep into it👇
bytebrief.vercel.app/blog/ge…
#GemmaScope2 #MechanisticInterpretability #AISafety #DeepMind #Gemma3 #SparseAutoencoders #Transcoders #OpenSourceAI #LLM #AIResearch #ModelInterpretability #GenAI
1
2
39

11 Dec 2025
Circuits, Features, and Heuristics in Molecular Transformers
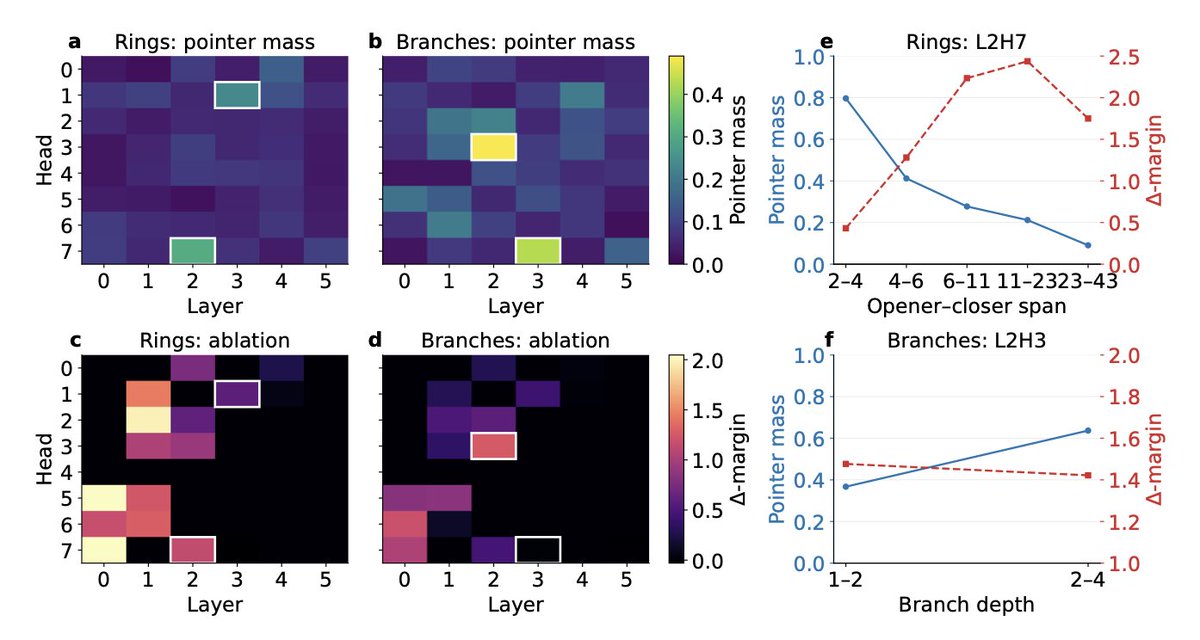
1. This study provides a deep dive into how molecular transformers generate valid and diverse chemical structures. The authors uncover the computational mechanisms that enable these models to capture the rules of molecular representation, revealing specialized attention heads and circuits that handle syntactic parsing and chemical validity constraints.
2. A key innovation is the identification of specific attention heads in transformers that act as "pointer circuits" for matching ring closures and balancing branches in SMILES strings. These heads are crucial for maintaining syntactic correctness during molecular generation.
3. The researchers also discover a distributed linear representation of valence capacity in the model's residual stream. By intervening on this direction, they can modulate bond-order predictions in a chemically consistent way, demonstrating the model's internal handling of chemical rules.
4. Using sparse autoencoders (SAEs), the study extracts interpretable feature dictionaries that align with chemically meaningful activation patterns. This approach allows for the identification of features corresponding to functional groups and substructures, enhancing the model's transparency.
5. The practical applications of these findings are showcased through improved performance on downstream tasks like property prediction and activity cliffs. SAE-derived features outperform traditional fingerprints and supervised baselines, indicating their potential for real-world drug development.
6. The authors also demonstrate that SAE features can be used to steer molecular generation at inference time, increasing structural similarity and guiding samples toward desired regions of chemical space without retraining.
📜Paper: arxiv.org/abs/2512.09757
#MolecularTransformers #ComputationalChemistry #AIinDrugDiscovery #SparseAutoencoders
2
1,116
10 Dec 2025
Unveiling Latent Knowledge in Chemistry Language Models through Sparse Autoencoders
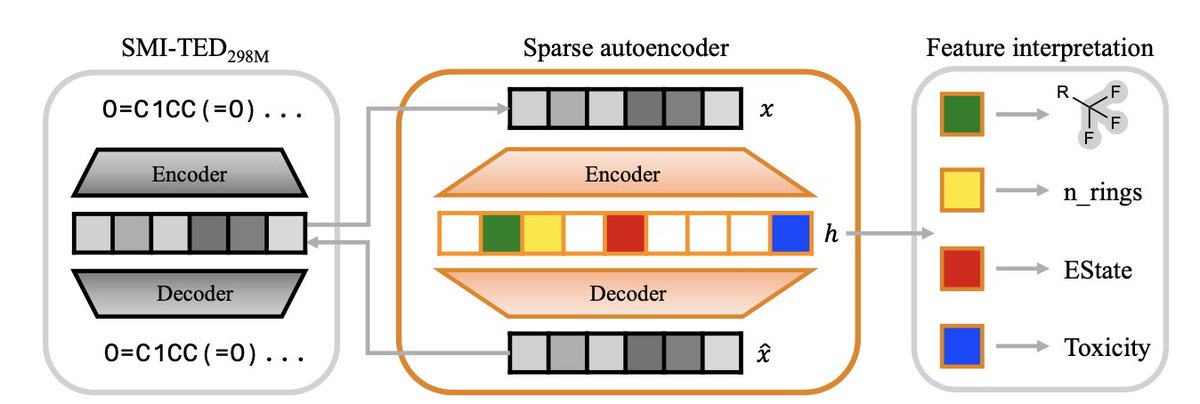
1. A novel study explores how sparse autoencoders (SAEs) can decode the latent knowledge within chemistry language models (CLMs), revealing interpretable chemical features and their activation patterns across diverse molecular datasets.
2. The research demonstrates that SAEs effectively disentangle complex molecular representations into interpretable features, identifying correlations between specific latent features and distinct domains of chemical knowledge, such as structural motifs, physicochemical properties, and pharmacological drug classes.
3. By applying SAEs to the state-of-the-art SMI-TED foundation model, the study extracts semantically meaningful latent features, providing a generalisable framework to uncover latent knowledge in chemistry-focused AI systems and accelerating computational chemistry.
4. The study shows that SAE features outperform individual neurons in detecting chemical concepts, especially for rare motifs, indicating the SAE constructs new detectors from linear combinations of neurons, a crucial step for capturing rare concepts not explicitly represented in the base model.
5. Feature steering experiments confirm the causal relationship between SAE features and their corresponding chemical motifs, enabling targeted modifications to molecular structures while preserving chemical validity, a capability not afforded by the original entangled neuron basis.
6. The research also highlights that SAE features provide a more disentangled representation of global physicochemical properties and higher-level functional concepts, such as toxicity prediction and pharmacological mechanisms, compared to raw neurons and standard dimensionality reduction techniques.
7. Limitations include the unscaled process of feature interpretation, confinement to a single model architecture, and the potential lack of generalisation to out-of-distribution molecules. Future work should focus on automated feature interpretation, AI safety applications, and exploring feature behaviour across different model architectures and scales.
📜Paper: arxiv.org/abs/2512.08077
#ChemistryLanguageModels #SparseAutoencoders #Interpretability #ComputationalChemistry #AIinScience
3
12
1,228
8 Dec 2025
Mechanistic Interpretability of Antibody Language Models Using SAEs
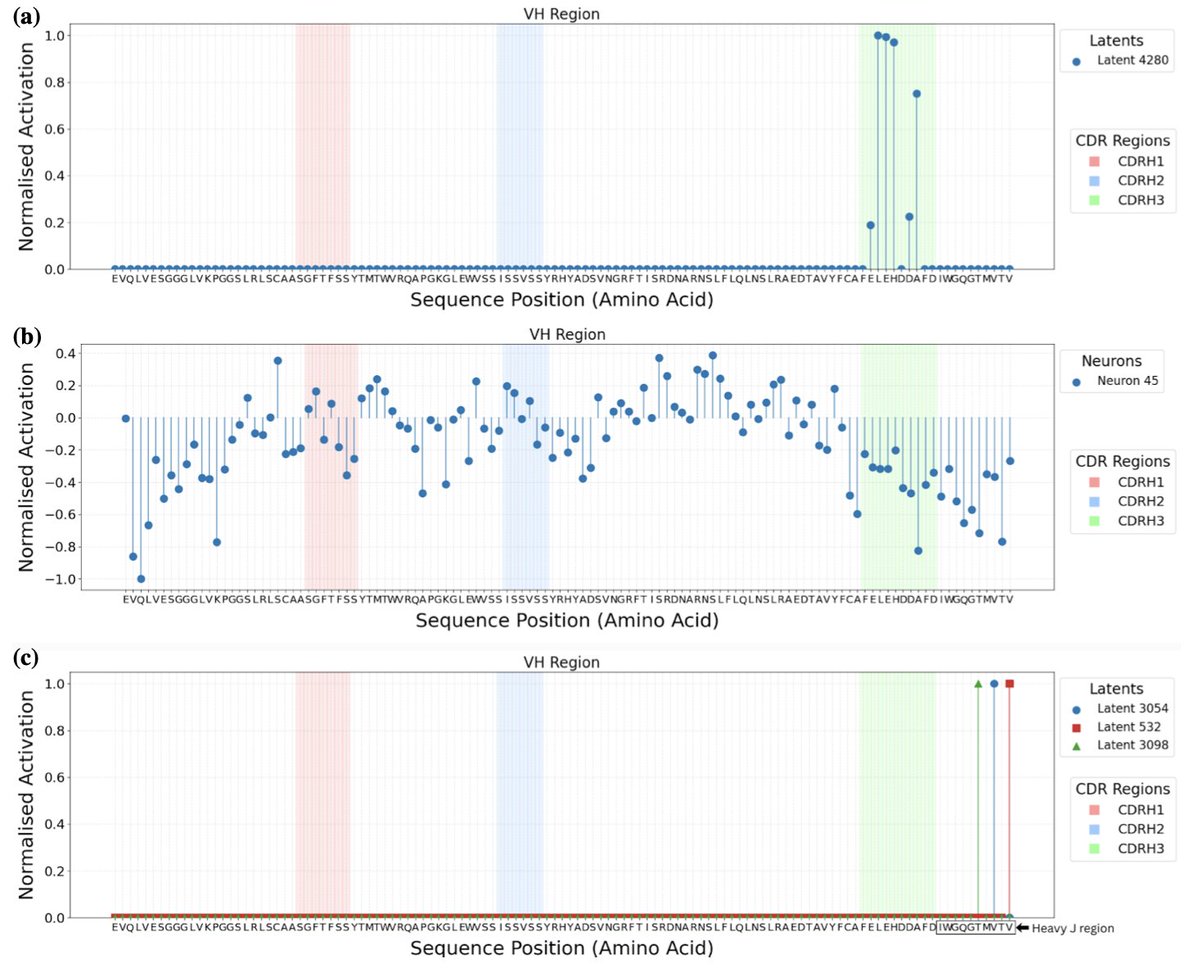
1. This study explores the use of Sparse Autoencoders (SAEs) to interpret and steer antibody language models, specifically focusing on the autoregressive model p-IgGen. It highlights how SAEs can uncover biologically meaningful features and their potential for controlling model generation.
2. The research employs TopK and Ordered SAEs to identify interpretable latent features in antibody models. TopK SAEs reveal meaningful biological concepts but struggle with causal control over generation, while Ordered SAEs provide more reliable steering at the cost of interpretability.
3. A key finding is that high correlation between features and concepts does not guarantee steerability. This insight is crucial for understanding the limitations of using SAEs for model manipulation in antibody design.
4. The study demonstrates that Ordered SAEs are more effective for steering model outputs, identifying hierarchical features that correspond to higher-level concepts rather than simple residue-level identities. This suggests a promising approach for generating antibody libraries with desired properties.
5. The work underscores the importance of mechanistic interpretability in protein language models, particularly for applications in drug discovery. It also highlights the need for more annotated datasets to enhance feature identification and steerability in antibody models.
📜Paper: arxiv.org/abs/2512.05794
#ComputationalBiology #AntibodyEngineering #SparseAutoencoders #ProteinLanguageModels #MechanisticInterpretability
1
7
33
2,421
1 Dec 2025
FoldSAE: Learning to Steer Protein Folding Through Sparse Representations
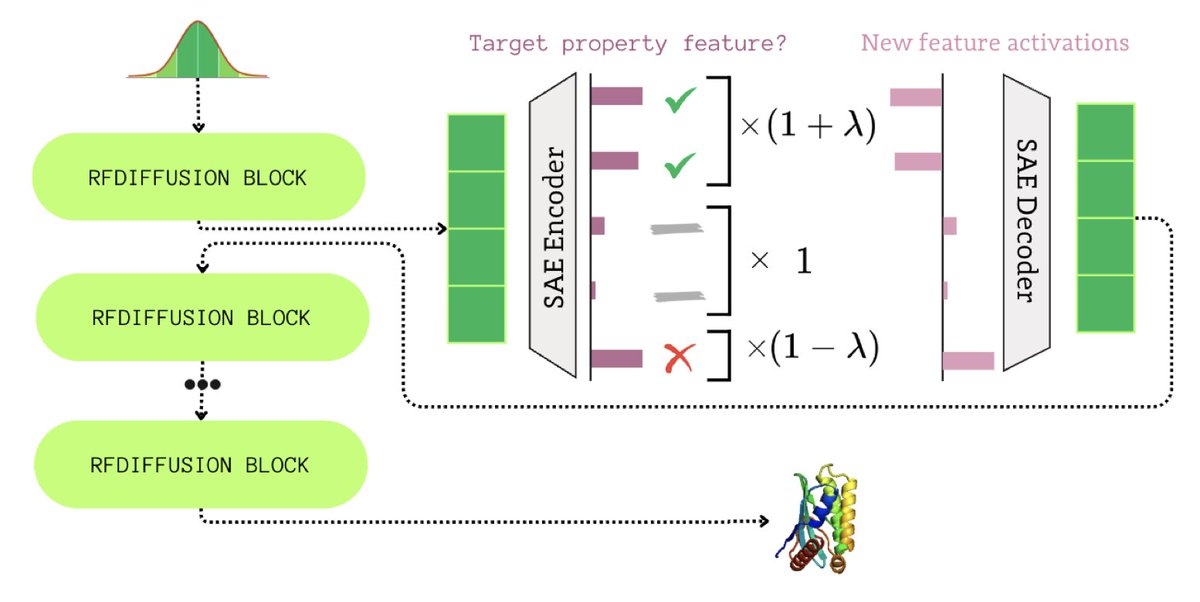
1. A groundbreaking study introduces FoldSAE, a novel framework that leverages Sparse Autoencoders to interpret and control protein folding within the RFdiffusion model. This work pioneers a method to make the black-box nature of protein structure generation more transparent and controllable.
2. The core innovation lies in decomposing RFdiffusion's dense internal representations into sparse, interpretable features using Sparse Autoencoders. This allows researchers to uncover how specific features correlate with secondary protein structures like helices and strands, offering unprecedented insight into the folding process.
3. A key finding is that certain latent features are antagonistic, positively correlating with helices while negatively correlating with strands. This dual role enables precise control over secondary structure formation through a tunable hyperparameter, steering the generation process towards desired structural outcomes.
4. The study validates its approach by demonstrating fine-grained control over protein secondary structures. By amplifying or suppressing specific features, researchers can significantly modulate the content of helices and strands in generated protein backbones, while maintaining biological plausibility.
5. FoldSAE not only enhances interpretability but also transforms protein design from stochastic sampling to precise engineering. Future work could extend this framework to control additional properties such as solvent accessibility and ligand binding, further advancing the field of computational protein design.
📜Paper: arxiv.org/abs/2511.22519v1
#ProteinFolding #SparseAutoencoders #ComputationalBiology #Interpretability #ProteinDesign
5
30
2,340
26 Oct 2025
Sparse Autoencoders Reveal Interpretable Features in Single-Cell Foundation Models
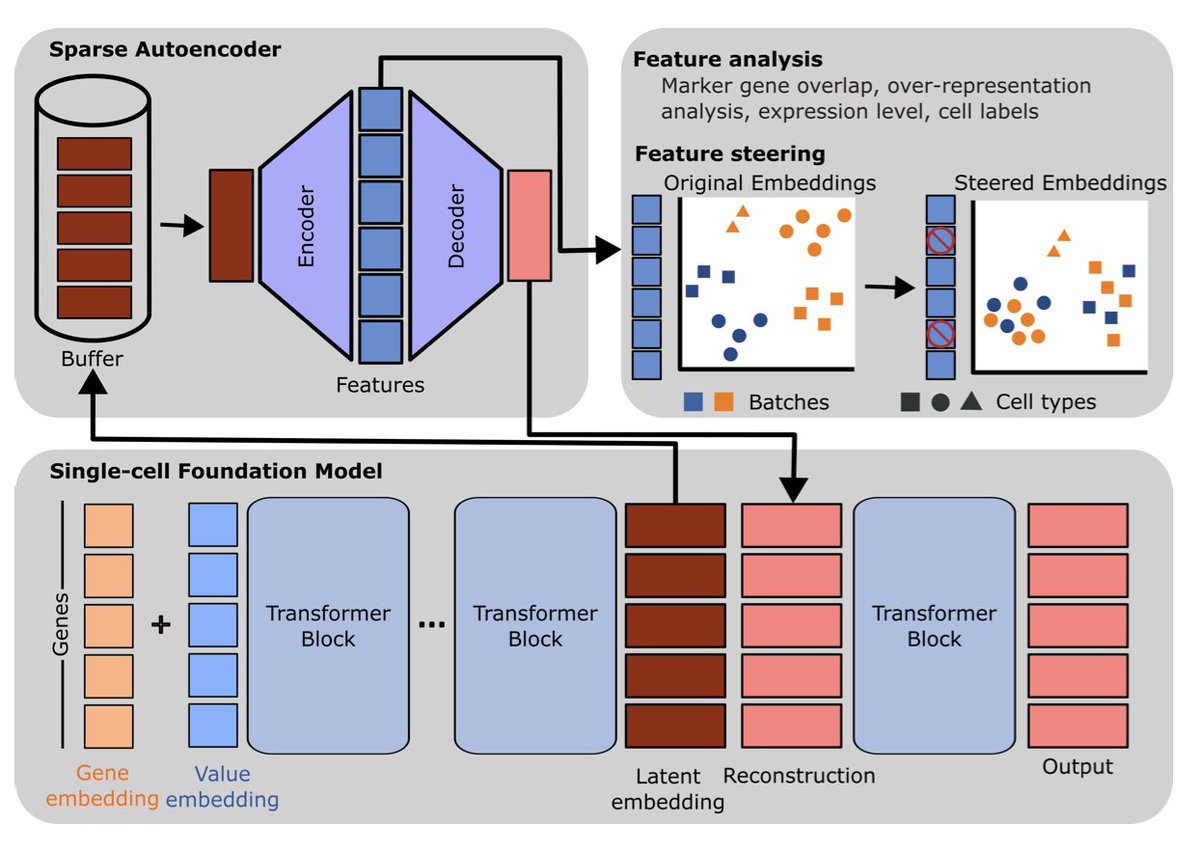
1. A new study by Pedrocchi et al. explores how sparse autoencoders (SAEs) can uncover meaningful biological and technical signals in single-cell foundation models (scFMs). This work provides insights into the complex representations learned by scFMs, which are crucial for applications like cell type annotation and data integration.
2. The researchers trained SAEs on the hidden representations of two widely used scFMs, scGPT and scFoundation. They found that these models capture diverse biological concepts, even in their pre-trained states, revealing both gene-specific and cell-specific features.
3. An important finding is that different training protocols and architectures of scFMs lead to distinct encodings of information. This suggests that the internal mechanisms of scFMs are highly influenced by their training procedures.
4. The study also highlights that while scFMs can capture cell type information across multiple studies, they often fail to unify this information into a single generalized representation. This limitation affects the models' generalizability across different datasets.
5. A novel contribution is the demonstration that SAE-derived features are causally related to model behavior. The authors show that by manipulating these features, they can reduce unwanted technical effects and improve batch integration in scFMs.
6. The work provides a path toward more interpretable and controllable single-cell foundation models. By understanding and steering the internal representations, researchers can potentially enhance the performance and usability of these models in various biological applications.
📜Paper: biorxiv.org/content/10.1101/…
#SingleCellRNASeq #SparseAutoencoders #InterpretableAI #ComputationalBiology #FoundationModels
1
4
19
1,722
5 Oct 2025
Towards Functional Annotation with Latent Protein Language Model Features
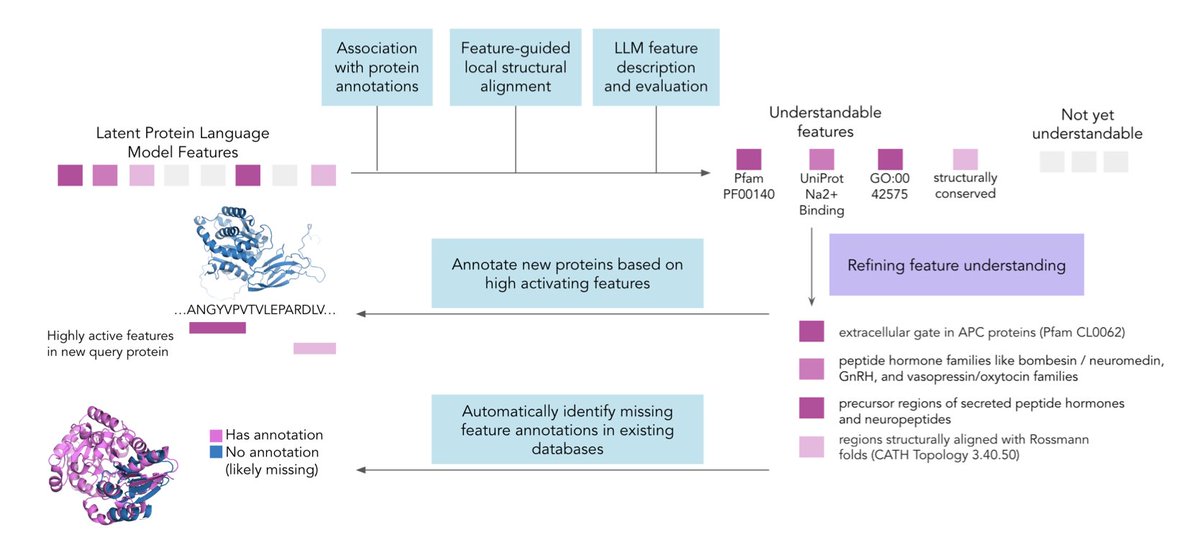
1. This study introduces a novel validation pipeline for functional protein annotation using latent features from protein language models (PLMs). The pipeline combines expanded database matching, feature-guided local structural alignment, and LLM-based feature description generation to identify cohesive and reliable features for annotation.
2. The researchers demonstrate that Sparse Autoencoder (SAE) features derived from PLMs can capture more granular patterns than existing protein databases, enabling the identification of sub-domains within proteins. This granular approach allows for the detection of missing annotations by identifying proteins with recognizable structural motifs that lack corresponding database labels.
3. The study shows that SAE features maintain structural consistency across unseen proteins, allowing for rapid annotation of metagenomic proteins. The pipeline identifies 615 structurally similar SAE features in unannotated metagenomic proteins, enabling the structural matching of at least 8,077 metagenomic proteins to characterized proteins.
4. The validation pipeline doubles annotation coverage, identifying over 60% of features with strong correspondence to biological annotations (F1>0.8). The integration of local structural alignment and LLM-based pattern recognition identifies additional features missed by existing codes, enhancing the practical applications of SAE features for protein annotation.
5. The study highlights the potential of using PLM SAE features for zero-shot generalization to novel metagenomic proteins, including those without matches to existing protein families. This approach offers a rapid annotation pipeline with constant time search regardless of database size, providing structural and functional information about the feature that triggered the match.
6. The researchers also demonstrate the ability to retrieve literature discussing specific protein regions where features activate, bridging the gap between feature identification and functional interpretation. This approach successfully identifies papers discussing exact regions for several features, revealing detailed functional insights.
7. The study concludes that while there are limitations, such as the need for manual validation of feature activations and potential misses in structural validation, the framework provides a useful proof-of-concept for advancing protein annotation using latent features from PLMs. Future work includes expanding analysis across multiple layers within PLMs and benchmarking against existing annotation approaches.
💻Code: github.com/jsilbergDS/toward…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinAnnotation #ProteinLanguageModels #SparseAutoencoders #Metagenomics #StructuralBiology #FunctionalGenomics
3
26
1,968
9 Sep 2025
ProtSAE: Disentangling and Interpreting Protein Language Models via Semantically-Guided Sparse Autoencoders
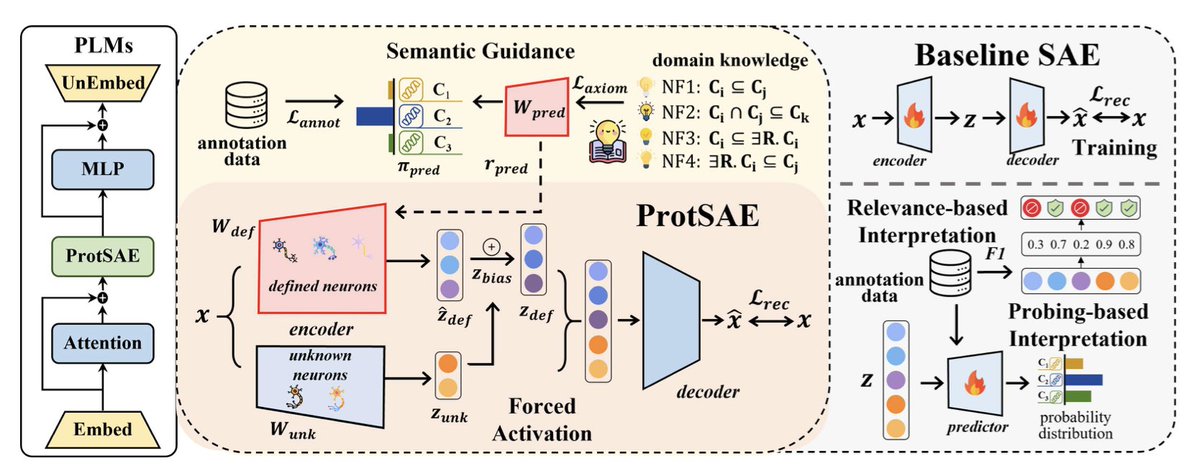
1. A novel method called ProtSAE has been proposed to improve the interpretability of protein language models (PLMs) by using semantically-guided sparse autoencoders. Unlike traditional sparse autoencoders (SAEs), ProtSAE incorporates semantic guidance during training to disentangle complex protein features and align them more closely with biological concepts.
2. ProtSAE introduces protein domain knowledge into the training process, leveraging ELEmbeddings to model logical constraints among concepts such as subsumption and conjunction. This enhances the interpretability and semantic consistency of the learned features, allowing for more accurate representation of biological concepts in the latent space of PLMs.
3. The method applies forced activations and feature rescaling to ensure that defined activations effectively participate in reconstruction with high fidelity. This is crucial for maintaining the biological relevance of the learned features while preserving the reconstruction quality of the model.
4. Extensive experiments demonstrate that ProtSAE captures more interpretable features that are closely aligned with biological concepts compared to previous methods. It consistently outperforms baselines across varying levels of sparsity while maintaining high reconstruction fidelity, showing its superiority in both interpretability and performance.
5. Steering experiments reveal that the semantic features learned by ProtSAE can effectively guide PLM outputs toward desired functional outcomes. This validates the quality of the learned representations and their potential for precise model control, opening up new possibilities for protein engineering and biological research.
📜Paper: arxiv.org/abs/2509.05309v1
#ProteinLanguageModels #SparseAutoencoders #Interpretability #Bioinformatics #ComputationalBiology
2
21
2,267
27 Aug 2025
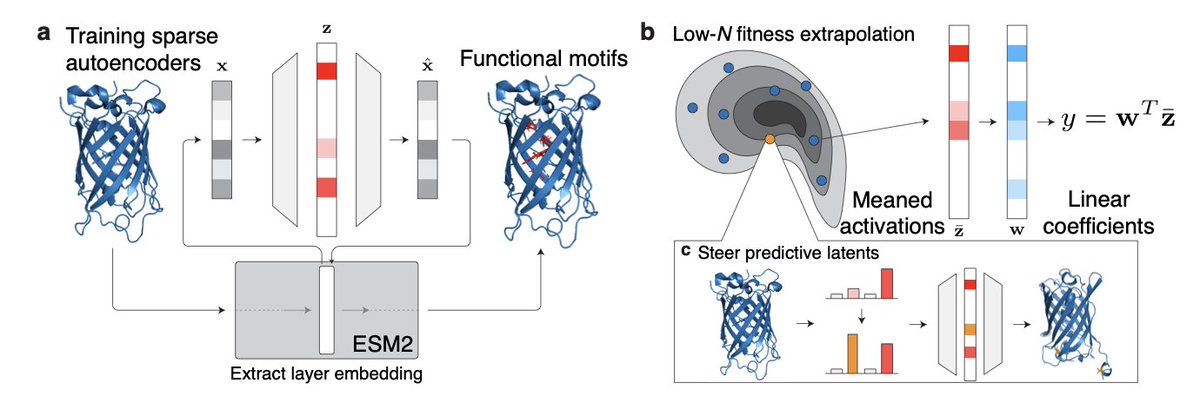
Sparse Autoencoders for Low-N Protein Function Prediction and Design
1. This study explores the use of Sparse Autoencoders (SAEs) for predicting protein function and designing proteins in low-data scenarios, demonstrating significant improvements over existing methods.
2. SAEs trained on fine-tuned ESM2 embeddings consistently outperform ESM2 baselines in fitness prediction tasks, even with as few as 24 sequences, showing their effectiveness in capturing biologically meaningful representations.
3. The study introduces a method to steer predictive latents in SAEs to design high-functioning protein variants, achieving top-fitness variants in 83% of cases compared to designing with ESM2 alone.
4. The authors analyze the best-performing variants in green fluorescent protein (GFP) and the IgG-binding domain of protein G (GB1), uncovering biologically meaningful motifs exploited by SAEs for steering.
5. SAEs achieve higher generalization to unseen variants compared to ESM2 in various low-N fitness extrapolation tasks, including position, regime, and score extrapolation.
6. The study highlights the importance of sparsity in SAEs, which compresses biologically relevant information into a sparse latent space, enhancing performance in low-N regimes.
7. The authors suggest future work could involve expanding the design space by steering multiple latents at once and coupling SAE steering with physics-based tools to optimize for both function and stability.
📜Paper: arxiv.org/abs/2508.18567v1
💻Code: github.com/amirgroup-codes/L…
#ProteinEngineering #MachineLearning #SparseAutoencoders #ProteinDesign #LowDataScenarios
2
54
4,933
20 Aug 2025
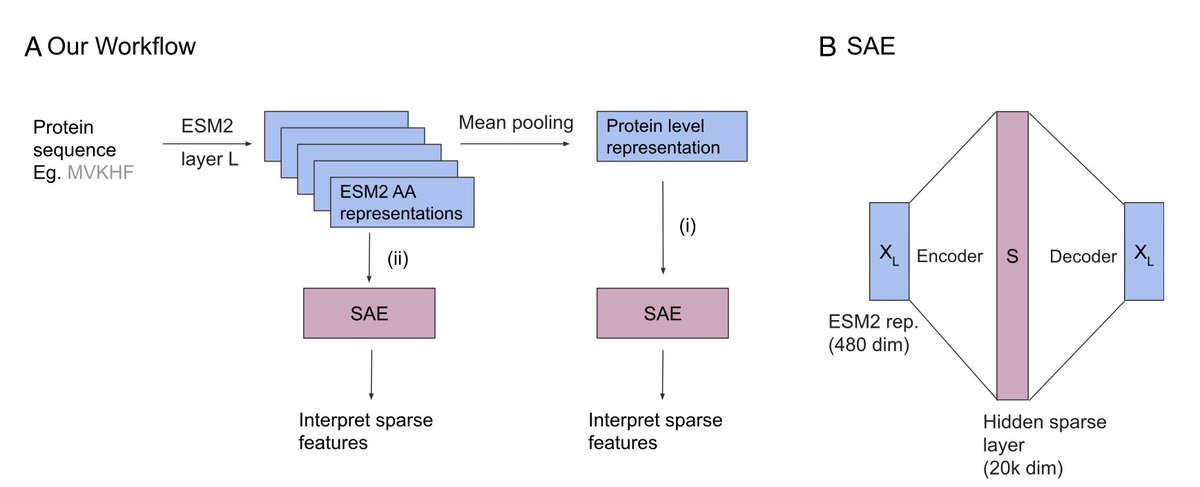
Sparse autoencoders uncover biologically interpretable features in protein language model representations @PNASNews
1. A groundbreaking study by Gujral et al. leverages sparse autoencoders (SAEs) and transcoders to extract interpretable features from protein language models (PLMs) in an unsupervised manner, enhancing the transparency and explainability of these models.
2. The research focuses on ESM2, a popular PLM, and demonstrates that SAEs can identify sparse features tightly associated with Gene Ontology (GO) terms across various levels of the GO hierarchy, revealing specific protein families and functions.
3. The study employs automated interpretation using Anthropic’s Claude, which finds that many extracted features correspond to distinct biological entities such as the NAD Kinase family and proteins involved in methyltransferase activity, showcasing the potential for biological insights.
4. A key innovation is the unsupervised extraction of features, which contrasts with traditional supervised methods, allowing for the discovery of biologically relevant information without prior knowledge.
5. The findings highlight that sparse features are more interpretable than standard ESM2 neurons, suggesting that SAEs and transcoders can significantly improve the understanding of PLM representations and their applications in downstream tasks.
6. This work not only enhances the interpretability of PLMs but also opens new avenues for extracting meaningful biological insights, fostering human-AI collaboration, and ensuring the safety and trustworthiness of these models.
📜Paper: pnas.org/doi/10.1073/pnas.25…
#ProteinLanguageModels #SparseAutoencoders #Bioinformatics #Interpretability #AIinBiology
8
42
2,468
18 Feb 2025
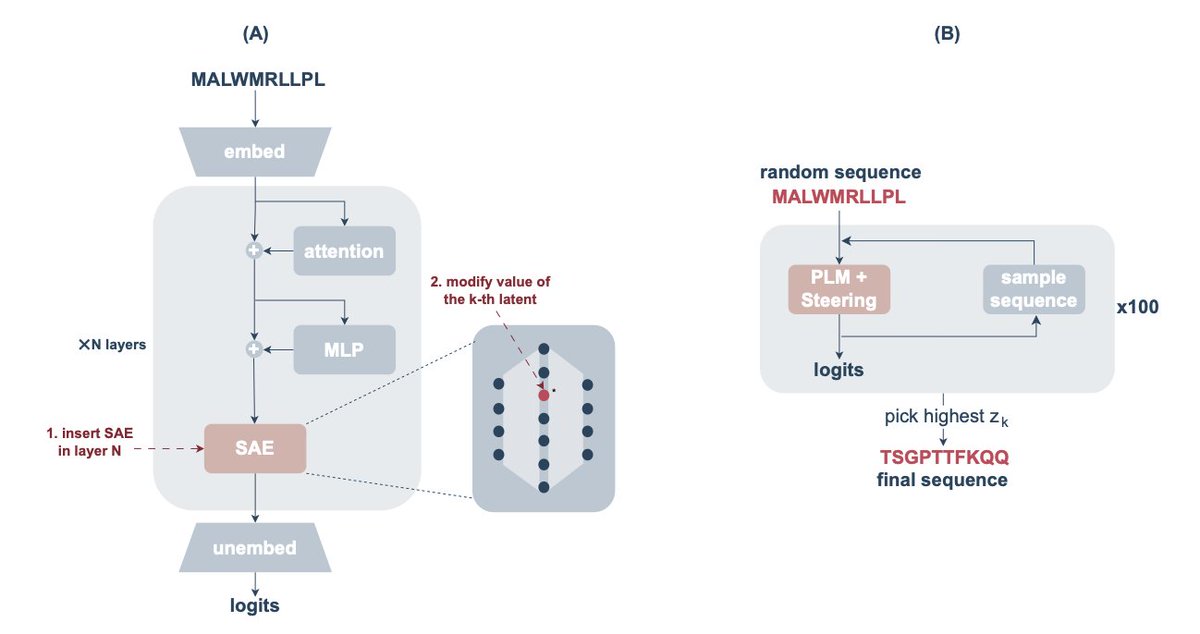
Interpreting and Steering Protein Language Models Through Sparse Autoencoders
1. This study introduces the use of sparse autoencoders (SAEs) to interpret protein language models like ESM-2, aiming to uncover meaningful latent features that represent specific biological characteristics such as binding sites and zinc finger motifs.
2. The authors propose a methodology for selecting layers from the transformer architecture of the ESM-2 model using an intrinsic dimension estimator to ensure that the most relevant abstract features are captured for interpretability.
3. By training the SAE on protein sequence data, the researchers successfully extracted and interpreted latent components associated with various functional features, demonstrating how these components can be leveraged for model steering.
4. A key innovation is the application of SAEs to steer the protein language model towards generating sequences with specific structural features like zinc fingers, showing the potential for generating complex protein sequences with desired properties.
5. The study provides a detailed evaluation of the precision and recall of latent-feature associations, revealing high-quality associations with functional features and introducing the concept of steering protein sequence generation through targeted latent manipulation.
6. The results showcase that the approach can generate functional protein sequences with zinc finger motifs, demonstrating the utility of sparse autoencoders for model steering in protein sequence design.
7. This work marks a significant advancement in the interpretability of protein language models, offering new insights into how these models encode biological information and providing a novel approach for protein design and engineering.
💻Code: github.com/edithvillegas/plm…
📜Paper: arxiv.org/abs/2502.09135
#ProteinLanguageModels #DeepLearning #Bioinformatics #ProteinDesign #MachineLearning #SparseAutoencoders #ZincFinger #SequenceDesign
1
1
16
2,014
18 Feb 2025
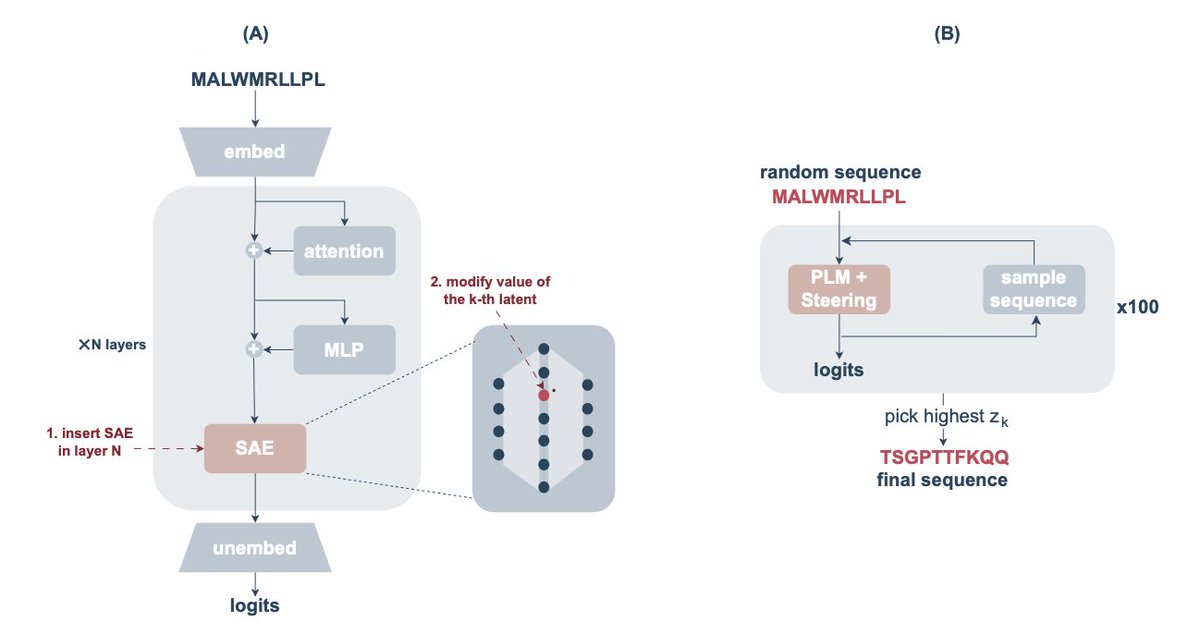
Interpreting and Steering Protein Language Models Through Sparse Autoencoders
1. This study introduces the use of sparse autoencoders (SAEs) to interpret protein language models like ESM-2, aiming to uncover meaningful latent features that represent specific biological characteristics such as binding sites and zinc finger motifs.
2. The authors propose a methodology for selecting layers from the transformer architecture of the ESM-2 model using an intrinsic dimension estimator to ensure that the most relevant abstract features are captured for interpretability.
3. By training the SAE on protein sequence data, the researchers successfully extracted and interpreted latent components associated with various functional features, demonstrating how these components can be leveraged for model steering.
4. A key innovation is the application of SAEs to steer the protein language model towards generating sequences with specific structural features like zinc fingers, showing the potential for generating complex protein sequences with desired properties.
5. The study provides a detailed evaluation of the precision and recall of latent-feature associations, revealing high-quality associations with functional features and introducing the concept of steering protein sequence generation through targeted latent manipulation.
6. The results showcase that the approach can generate functional protein sequences with zinc finger motifs, demonstrating the utility of sparse autoencoders for model steering in protein sequence design.
7. This work marks a significant advancement in the interpretability of protein language models, offering new insights into how these models encode biological information and providing a novel approach for protein design and engineering.
@ansuin
💻Code: github.com/edithvillegas/plm…
📜Paper: arxiv.org/abs/2502.09135
#ProteinLanguageModels #DeepLearning #Bioinformatics #ProteinDesign #MachineLearning #SparseAutoencoders #ZincFinger #SequenceDesign
1
5
17
1,612
18 Nov 2024
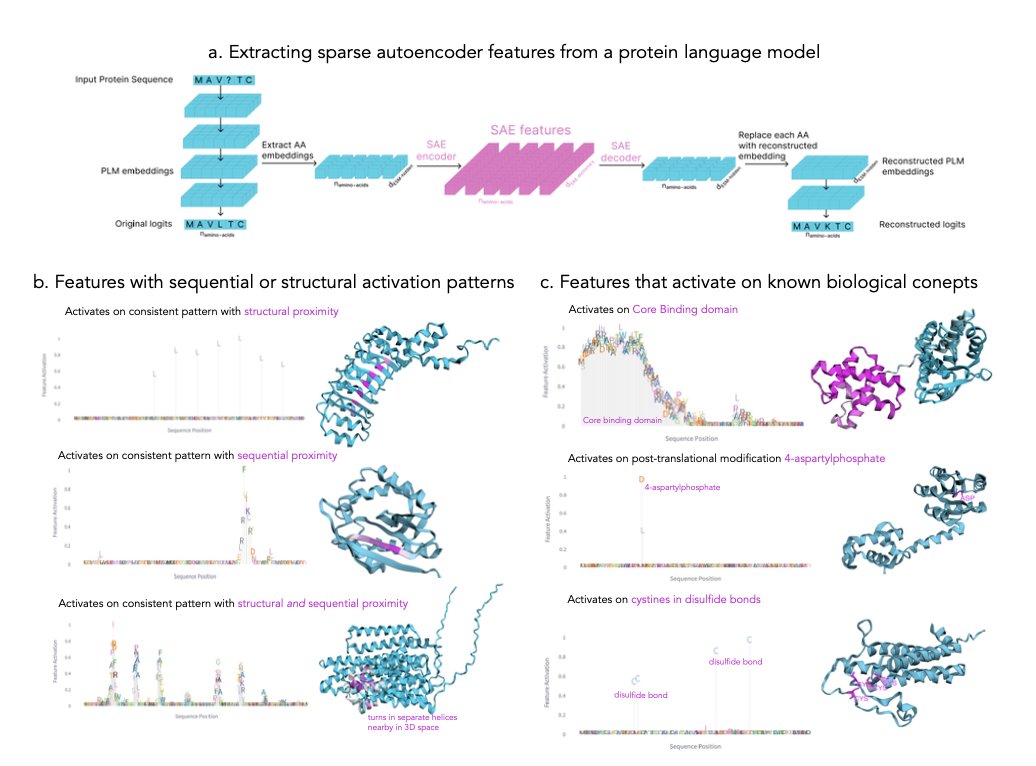
InterPLM: Discovering Interpretable Features in Protein Language Models via Sparse Autoencoders
• InterPLM introduces a novel framework to analyze protein language models (PLMs) using Sparse Autoencoders (SAEs), uncovering over 2,500 interpretable features per model layer.
• By examining embeddings from ESM-2, these features align with 143 biological concepts, including binding sites, structural motifs, and functional domains, far surpassing previous neuron-based approaches that identified only 15 concepts.
• A key innovation lies in uncovering features associated with novel, unannotated protein patterns, offering new directions for biological research and protein database enrichment.
• The InterPLM pipeline provides an interactive dashboard (interPLM.ai) for exploring feature activation patterns, enabling researchers to visualize sequential, structural, and functional relationships.
• The framework demonstrates practical applications such as annotating missing biological features in protein databases and steering protein sequence generation with targeted feature activation.
• Quantitative comparisons show that InterPLM features outperform raw neuron interpretations in biological relevance, offering a breakthrough in PLM interpretability.
• InterPLM also leverages large language models to generate automatic descriptions for unannotated features, achieving strong predictive power for protein activation levels.
• Future directions include applying the framework to structural prediction models like AlphaFold and expanding its capacity for feature-based steering in complex biological contexts.
@ElanaPearl @james_y_zou
💻Code: github.com/ElanaPearl/interP…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLanguageModels #AIforBiology #Bioinformatics #InterpretableAI #SparseAutoencoders
1
13
60
5,981

22 Jun 2024
New attention sparseautoencoders post dropped.
$1000 bounty to whomever find the best attention circuit!
21 Jun 2024

Great post from my scholars @Connor_Kissane
& @robertzzk!
SAEs are fashionable, but are they a useful tool for researchers? They are! We find a deeper understanding of the well-studied IOI circuit, and make a circuit analysis tool
$1000 bounty to whoever finds the best circuit!
1
5
2,307

8 Jun 2024
Deep Dive into Anthropic’s SparseAutoencoders by Hand
towardsdatascience.com/deep-…
@JagersbergKnut @chidambara09 @KevinClarity
@enilev @EstelaMandela @RLDI_Lamy
@Analytics_699 @_deus__machina @bimedotcom
@AndrewinContact
@HLStockenstrom @sulefati7 @sonu_monika
@Khulood_Almani
1
5
8
325