Diffusion-based generative model with scaffold-hopping strategy yields highly potent bioactive molecules
1 SMarT-Diff (Scaffold-based Multi-property Tuning Diffusion) is presented as a score-based diffusion framework for lead optimization that explicitly targets the classic tension between multi-objective property control and true scaffold-level exploration (scaffold hopping rather than close analog generation).
2 The most concrete outcome is wet-lab validation on LRRK2: three generated compounds were synthesized and tested (ADP-Glo Kinase Assay), and the best candidate lrrk2_m_1001 achieved IC50 = 1.544 nM, outperforming the positive control LRRK2-IN-1 (IC50 = 3.141 nM).
3 The core idea is to condition generation on Bemis–Murcko scaffolds as structural priors, while simultaneously guiding toward drug-likeness (QED), synthetic accessibility (SA), and pharmacophore matching; for CNS-relevant tasks, predicted BBB permeability is added as an extra objective.
4 Architecturally, SMarT-Diff uses a graph diffusion transformer (DiT) inside a score-based generative model (SGM), denoising unified graph tokens with adaptive layer normalization (AdaLN) to better encode topology and scaffold–substituent relationships.
5 Sampling is a two-level system: an inner Reverse Diffusion Predictor plus Adaptive Momentum Corrector (RA) improves stability and “chemotype fidelity,” while an outer Advantage Actor-Critic (A2C) loop steers sampling using pharmacophore-matching rewards and explicitly penalizes excessive scaffold similarity to stay in a scaffold-hopping regime.
6 Ablations on LRRK2 dissect the trade-offs: scaffold-graph conditioning restores high validity (to ~0.953) and boosts success rate; RA increases scaffold similarity (up to ~0.732), while A2C is used to pull similarity down toward the intended hopping window (centered near ~0.36–0.41) without collapsing diversity.
7 Scaffold-level out-of-distribution (OOD) generation is quantified at two abstraction levels: among 10,000 LRRK2 designs, 93.96% had Bemis–Murcko scaffolds unseen in training and 60.08% had novel generic scaffolds; nearest-neighbor scaffold similarity is centered around ~0.4 vs training, aligning with a practical scaffold-hopping definition.
8 Importantly, novelty is not treated as a pure exploration metric: within a filtered “drug-like and strong docking” subset (e.g., Glide SP < −8.0 kcal/mol, QED > 0.6, SA > 0.6), molecules with similarity < 0.5 still retain strong predicted binding (median Glide SP ≈ −8.57 kcal/mol), suggesting scaffold changes can preserve affinity.
9 Against scaffold-hopping baselines on LRRK2 (PMDM, DECOMPOPT, DRLinker, Tree-Invent, TurboHopp, DiffHopp), SMarT-Diff reports a balanced profile: novelty 1.000, validity 0.944, uniqueness 0.851, scaffold similarity 0.362 with diversity 0.749, top QED (0.640), strong SA (0.653), and the best success rate (0.629 with QED > 0.4 and SA > 0.6).
10 Beyond single-target kinase optimization (LRRK2, HPK1), the framework is shown to generalize to a GPCR (GLP-1R) without retraining and to dual-target design (GSK3β/JNK3) via MCS-mined shared cores plus pharmacophore matching, yielding candidates with favorable docking distributions and MM/GBSA support for dual-pocket stability.
📜Paper: doi.org/10.1002/advs.75674
#DrugDiscovery #GenerativeAI #DiffusionModels #MolecularGeneration #ScaffoldHopping #LeadOptimization #ComputationalChemistry #Cheminformatics #ReinforcementLearning #KinaseInhibitors

8
51
2,289

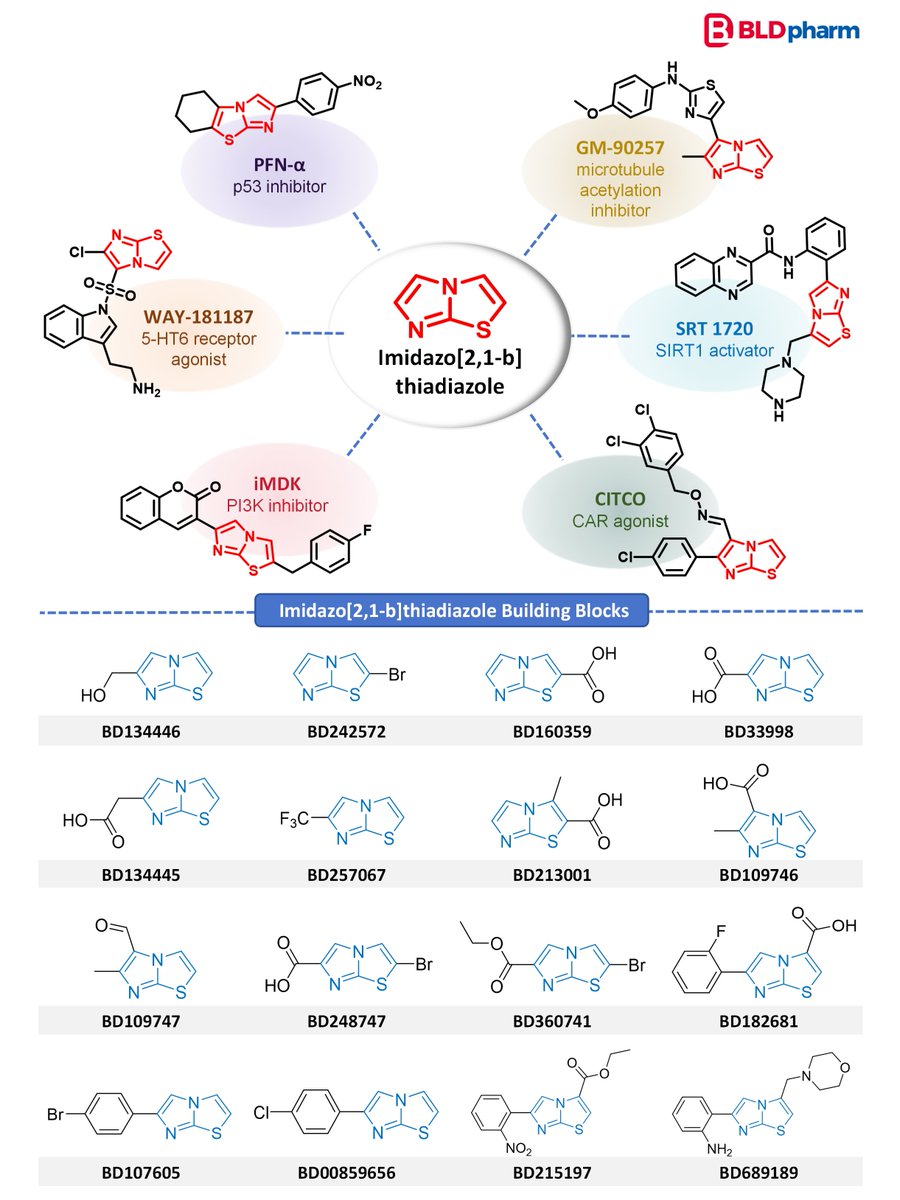
#BLDSeries | Imidazo[2,1-b]thiadiazole in Bioactive Molecules
Fused heterocycles continue to play an important role in medicinal chemistry. Imidazo[2,1-b]thiadiazole is a compact bicyclic scaffold with a heteroatom-rich framework, offering versatile electronic properties and diverse interaction patterns with biological targets.
This motif has appeared in a variety of bioactive molecules, including:
• p53 inhibitors
• PI3K inhibitors
• Microtubule acetylation inhibitors
• SIRT1 activators
• CAR agonists
• 5-HT6 receptor agonists
These examples highlight the value of imidazo-thiadiazole scaffolds as versatile heterocycles in drug discovery.
Relevant building blocks from @BldPharm:
bldpharm.com/search/BatchSea…
#MedicinalChemistry #DrugDiscovery #HeterocyclicChemistry #ScaffoldDesign #BuildingBlocks #LeadOptimization
1
2
25
841
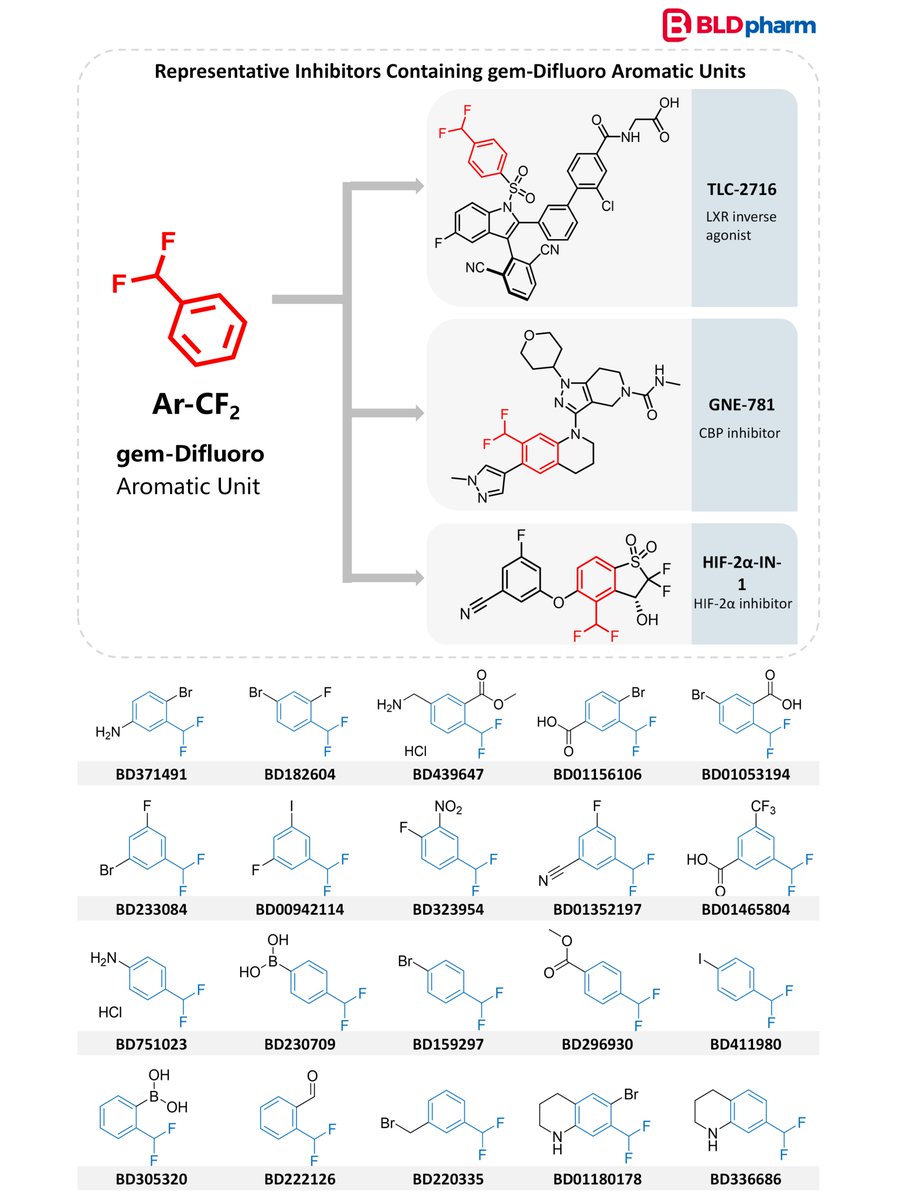
#BLDSeries | gem-Difluoro Aromatic Units: Electronic Modulation in Drug Design
Fluorine remains one of the most powerful tools for tuning molecular properties in medicinal chemistry. gem-Difluoro substitution on aromatic rings introduces a strong inductive effect that reshapes local electronic environments while maintaining compact steric profiles. This modification can influence aromatic electron density, adjust the acidity or basicity of nearby functional groups, and subtly modulate molecular polarity.
Relevant building blocks from @BldPharm :
bldpharm.com/search/BatchSea…
For inquiries, feel free to contact sales@bldpharm.com.
#MedicinalChemistry #DrugDiscovery #FluorineChemistry #ElectronicEffects #BuildingBlocks #LeadOptimization #ChemicalTools #TLC2716 #GNE781
2
18
882
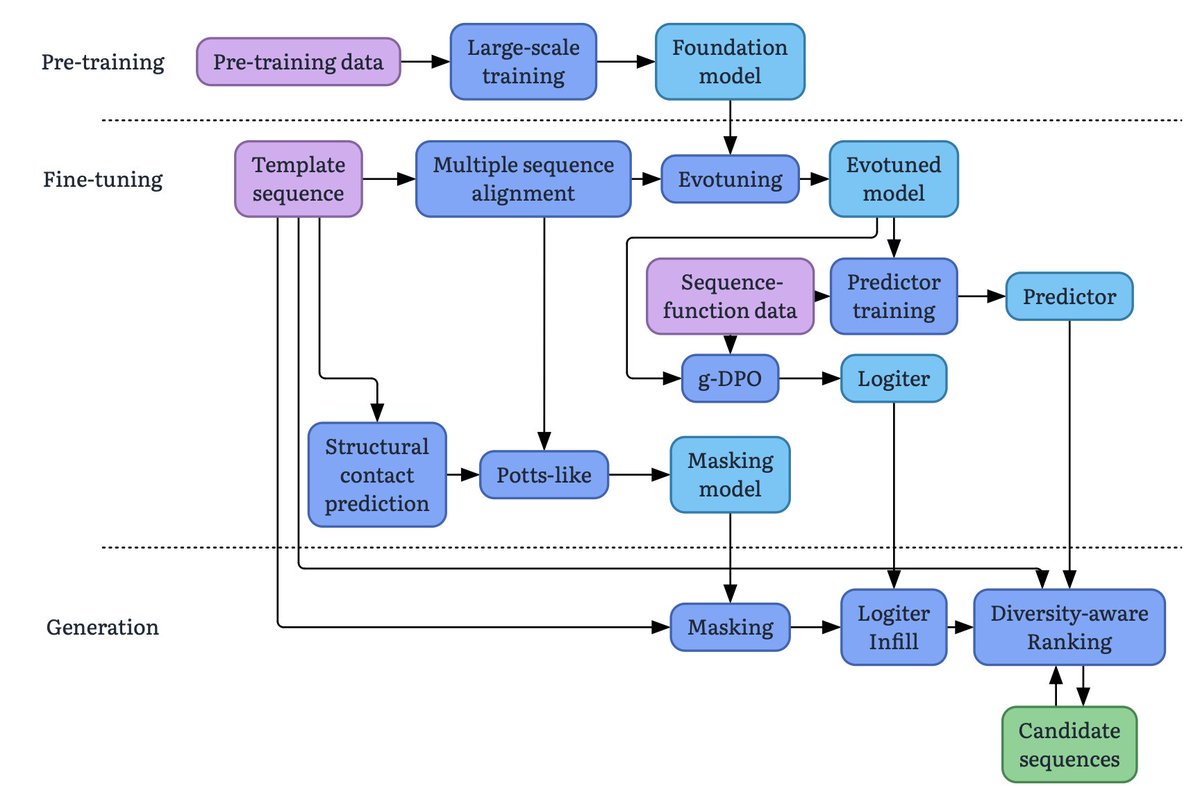
What comes after de novo? Automated lead optimization of proteins with CRADLE-1
1. CRADLE-1 is an automated machine learning framework for protein lead optimization that achieves 4-7x speedup compared to rational design, reducing wet lab rounds from months to days across diverse modalities including VHHs, scFvs, IgGs, peptides, enzymes, CRISPR systems, and vaccines.
2. The system uniquely enables multi-property optimization (1-6 properties simultaneously, up to 8 in private benchmarks) including binding affinity down to picomolar levels, thermostability, expression, activity, aggregation, nonspecificity, and immunogenicity.
3. Unlike structure-based de novo design methods, CRADLE-1 uses protein language models fine-tuned through three stages: unsupervised evotuning on evolutionary neighborhoods, supervised preference optimization via g-DPO, and regression-based property prediction, allowing black-box consumption of wet lab data without mechanistic knowledge.
4. The framework demonstrates remarkable data efficiency—achieving reliable optimization with as few as 12 sequences in zero-shot settings and typically requiring only 96-well plates per round, making it accessible for resource-constrained campaigns.
5. Key technical innovations include automated batch effect robustness, multi-property Spearman rank correlation for model evaluation, and a double-beam search generation strategy that maintains diversity while exploring high-function candidates.
6. Validation across 10 case studies shows consistent outperformance of baselines: winning the Adaptyv EGFR competition with 339 pM binders, improving P450 enzyme activity 40.6-fold versus 17.9-fold via rational design, and rescuing previously failed IgG and peptide optimization campaigns for top-20 pharmaceutical partners.
7. The system achieves 90-95% success rate compared to 85% industry standard for lead optimization, with built-in "optimization headroom" estimation to help teams avoid sunk cost fallacy by quantifying predicted improvement potential before committing resources.
8. CRADLE-1 operates as a fully automated API or UI service—users input template sequences and assay data, receiving designed sequences within approximately two GPU-days of compute (hours wall-clock with parallelization), without requiring structural data or biochemical expertise.
📜Paper: biorxiv.org/content/10.64898…
#CRADLE1 #ProteinEngineering #LeadOptimization #ProteinLanguageModels #MachineLearning #DrugDiscovery #AntibodyDesign #EnzymeEngineering #CRISPR #VaccineDesign
4
37
6,183
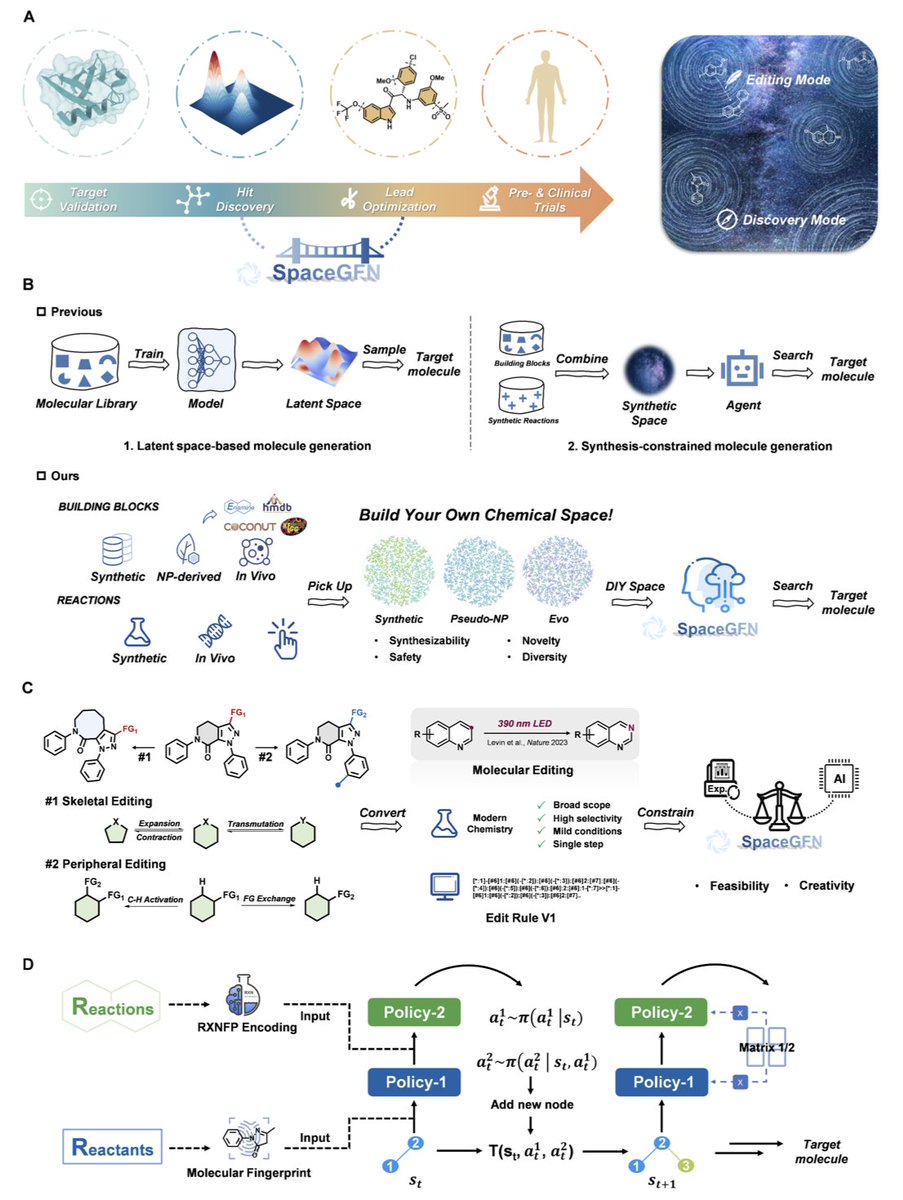
Designing the Haystack: Programmable Chemical Space for Generative Molecular Discovery
1. The authors introduce SpaceGFN, a generative framework that treats chemical space as a programmable computational object rather than a fixed distribution, enabling researchers to explicitly design and navigate molecular universes tailored to therapeutic hypotheses.
2. The framework operates in two modes: Discovery mode for de novo molecular generation and Editing mode for lead optimization, both built upon Generative Flow Networks (GFlowNets) that guarantee synthetic feasibility through reaction-defined construction.
3. In Discovery mode, SpaceGFN implements a DIY chemical space framework where users can combine custom building block libraries with reaction libraries. The paper demonstrates two innovative spaces: Pseudo-NP (pseudo-natural product space) constructed from NP-derived fragments, and Evo space built from endogenous metabolites and enzymatic reactions.
4. The Evo space represents a particularly compelling innovation—it embeds evolutionary biochemistry as a structural prior, biasing generated molecules toward favorable ADMET properties from the outset rather than relying solely on post-hoc filtering with limited prediction models.
5. The Evo space significantly outperforms synthetic controls across 28 of 35 ADMET properties, showing particular advantages in metabolism and toxicity profiles, though with higher polarity (TPSA) that presents both challenges and opportunities for formulation strategies.
6. In Editing mode, SpaceGFN introduces molecular editing into generative AI through the curated Edit Rule V1 dataset containing 300 reaction templates spanning single-atom editing, multi-atom editing, C-H activation, and functional group exchange.
7. This editing approach enables "digital medicinal chemistry" where each optimization step corresponds to an executable synthetic transformation, providing explicit synthetic routes rather than black-box modifications.
8. Large-scale validation across 96 diverse drug targets demonstrates robust optimization performance: 98.8% of targets showed improvement, with 84.2% exceeding 1 kcal/mol and 45.2% exceeding 2 kcal/mol in docking scores, while scaffold diversity increased 76% and topological diversity increased 98%.
9. The framework's modular architecture allows plug-and-play incorporation of new editing methodologies as they emerge from the synthetic chemistry literature, making it an evolving platform rather than a static tool.
10. By decoupling space definition from exploration, SpaceGFN returns design autonomy to experimental scientists while maintaining the efficiency advantages of generative AI, establishing a paradigm shift from "finding needles in haystacks" to "designing better haystacks."
💻Code: github.com/SpaceGFN/SpaceGFN
📜Paper:arxiv.org/abs/2603.00614
#DrugDiscovery #GenerativeAI #ChemicalSpace #MolecularDesign #GFlowNet #ADMET #SyntheticChemistry #LeadOptimization #NaturalProducts #Metabolites
1
5
31
1,807
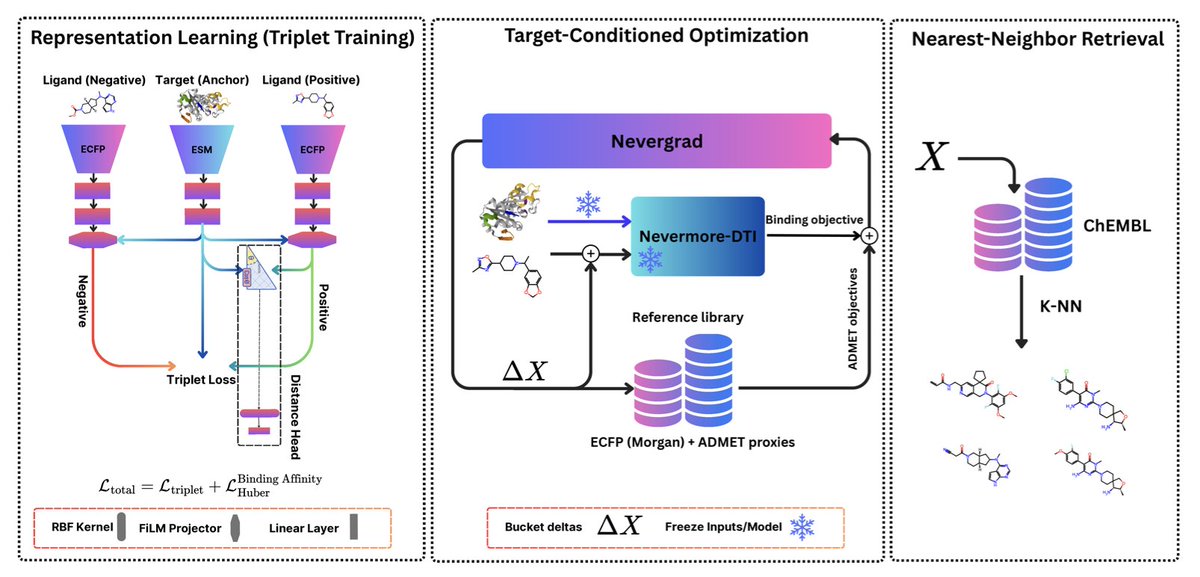
Nevermore: Target-Conditioned Protein–Ligand Representation Learning for Multi-Objective Lead Optimization with Database-Grounded Retrieval
1. Nevermore introduces a novel framework for multi-objective lead optimization that combines protein–ligand affinity prediction with ADMET constraints, leveraging a database-grounded approach to ensure chemical validity and interpretability.
2. The framework utilizes a geometry-aware protein–ligand affinity oracle, which aligns protein and ligand representations under contrastive objectives, improving over previous benchmarks and providing a stronger scoring signal for downstream optimization.
3. Nevermore optimizes in count-based Morgan fingerprint space, enabling discrete and interpretable edits to molecular structures. This approach allows for sparse, integer-constrained modifications that can be directly mapped to chemically meaningful substructures.
4. A key innovation is the use of Nevergrad for derivative-free optimization in fingerprint space, coupled with nearest-neighbor retrieval from a large compound library to convert optimized fingerprints into valid molecules without exhaustive enumeration.
5. Evaluations on Menin and SARS-CoV-2 Mpro targets demonstrate that Nevermore consistently retrieves candidate sets with improved affinity–property trade-offs compared to random sampling and similarity-based retrieval, maintaining explicit control and interpretability through discrete feature-space edits.
6. The study highlights the importance of balancing affinity optimization with ADMET constraints, showing that Nevermore can identify meaningful Pareto improvements even for challenging targets like Menin, which has a PPI-like pocket geometry.
7. Structural analysis of optimized ligands reveals that Nevermore introduces or strengthens multiple substructure motifs aligned with the intended edits, providing a plausible binding rationale and supporting the framework's ability to enhance interaction quality.
8. The approach is fast enough for large library settings and offers transparency by linking discrete fingerprint edits to specific chemical motifs, making it a practical tool for medicinal chemistry decision-making.
📜Paper: biorxiv.org/content/10.64898…
#Nevermore #LeadOptimization #ProteinLigand #MultiObjective #DatabaseGrounded #DrugDiscovery #ComputationalBiology
1
14
1,286

18 Dec 2025
AI is speeding up lead optimization with deep learning–driven suggestions. 📷📷
#AIDrugDiscovery #LeadOptimization #DeepLearning #MolecularDesign #MedicinalChemistry #AIinPharma #ComputationalChemistry #BiotechInnovation #LifeSciences #FutureOfMedicine
1
2
14

11 Dec 2025
Imagine you generated a hit that binds and inhibits your target with high potency, but in preclinical or clinical settings, it shows poor bioavailability or unexpected toxicity.
This is exactly where the Boltzmann AI Discovery Suite steps in!
In this clip, our SME Catherene Tomy shows how Boltzmann’s Molecule Optimization engine can take any structure and refine it for safer and stronger ADMET profiles, adjusting everything from carcinogenicity to metabolism to BBB permeability. From the same parent structure, the engine searches nearby chemical space. It optimizes only specific regions, rather than replacing the entire molecule, to identify non-carcinogenic and more drug-like variants.
You are not restricted to a single model. You can select any ADMET property you want to tune, including blood-brain barrier permeability, lipophilicity, half-life, clearance, and metabolism!
A fast look at how AI can turn a promising hit into a viable lead.
Discover the full Boltzmann AI Discovery Suite to see how far your research can go.
🔗boltzmann.co/oursuite
#DrugDiscovery #AIinBiotech #SmallMoleculeDesign #LeadOptimization #ADMET #ComputationalChemistry #AIDrugDiscovery #ChemistryInnovation #BoltzmannLabs
6
82

17 Oct 2025
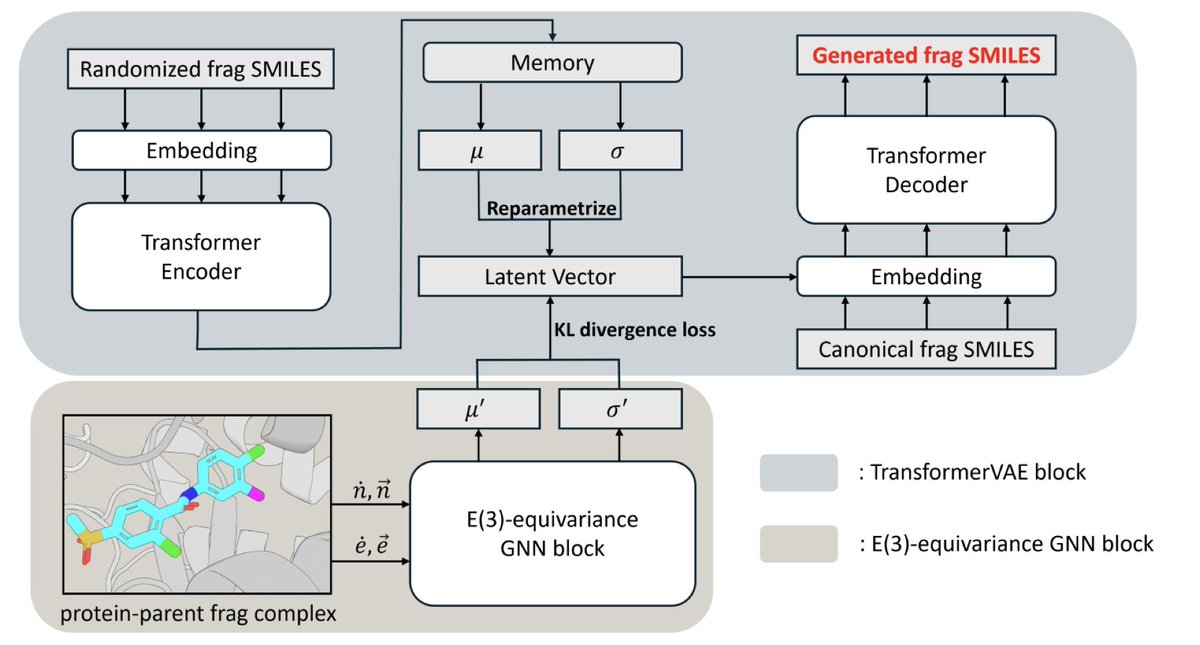
Slogen: A Structure-based Lead Optimization Model Unifying Fragment Generation and Screening
1. Slogen is a groundbreaking model in the field of structure-based drug design, aiming to enhance the efficiency and effectiveness of lead optimization. It combines fragment generation and screening in a unified framework, addressing key challenges in synthetic feasibility and structural innovation.
2. The model integrates a transformer-based variational autoencoder (VAE) with an E(3)-equivariant graph neural network (GNN). The VAE is pretrained on a diverse set of fragments, enabling both generative decoding and similarity-based screening. The GNN captures 3D protein–fragment interactions, predicting optimal fragment elaborations.
3. Slogen demonstrates superior performance in fragment elaboration tasks, achieving higher hit rates and better binding affinities compared to state-of-the-art models like Delete and DeepFrag. It also explores a broader chemical space, generating more diverse and drug-like molecules.
4. In screening tasks, Slogen shows competitive performance, particularly on the Delete test set, significantly outperforming traditional methods such as AutoDock Vina. This highlights its potential for practical applications in fragment-based drug discovery.
5. Case studies on the Smoothened receptor (SMO) and D1 dopamine receptor (D1DR) further illustrate Slogen's ability to design high-affinity, drug-like molecules. These examples demonstrate its versatility and practical utility in real-world drug design scenarios.
6. Despite its strengths, Slogen has areas for improvement. It performs well in generating chemically plausible ring structures but struggles with capturing the distribution of BM scaffolds. Future work could focus on pretraining with a more diverse fragment set and incorporating advanced GNN architectures.
7. The study emphasizes the importance of a balanced fragmentation strategy that ensures both synthetic tractability and chemical diversity. Slogen's unified approach provides a scalable route toward structure-guided lead optimization, bridging the gap between fragment generation and screening.
📜Paper: biorxiv.org/content/10.1101/…
#Slogen #LeadOptimization #StructureBasedDesign #DrugDiscovery #AIinPharma #FragmentGeneration #Screening #ComputationalBiology
2
13
1,380
3 Oct 2025
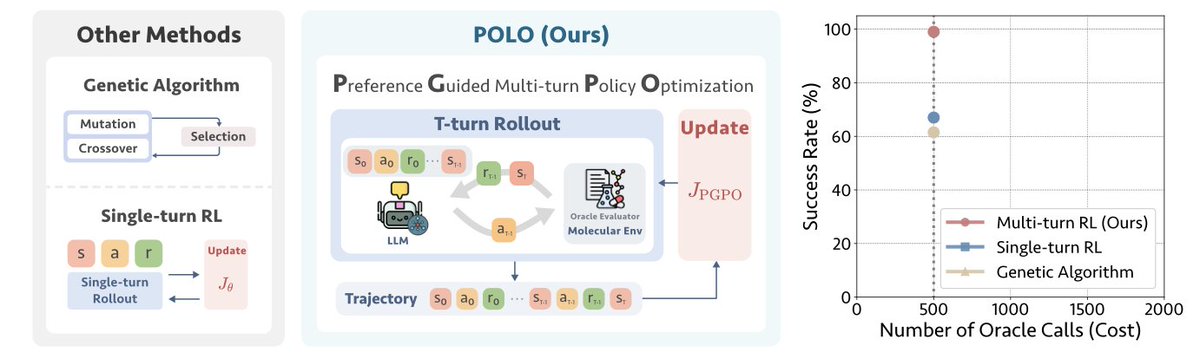
Lead Optimization with POLO: A New Benchmark in Sample Efficiency
1. The article introduces POLO (Preference-guided multi-turn Optimization for Lead Optimization), a novel framework that leverages multi-turn reinforcement learning to enhance molecular optimization in drug discovery. POLO achieves remarkable sample efficiency by learning from complete optimization trajectories rather than isolated steps, making it a game-changer in the field.
2. POLO’s core innovation is the Preference-Guided Policy Optimization (PGPO) algorithm. This algorithm combines trajectory-level optimization with turn-level preference learning, extracting dense comparative feedback from intermediate steps to reinforce successful strategies. This dual-level learning significantly improves sample efficiency, allowing POLO to achieve superior performance with limited oracle evaluations.
3. In experiments, POLO demonstrates an average success rate of 84% on single-property tasks and 50% on multi-property tasks using only 500 oracle evaluations. This represents a 2.3× improvement over baselines on single-property tasks and a significant advancement in sample-efficient molecular optimization.
4. POLO’s multi-turn approach aligns with the iterative nature of lead optimization, enabling the model to build on previous attempts and develop long-term strategies. This is a major departure from traditional methods that treat each optimization step independently, often discarding valuable historical information.
5. The framework includes critical supporting components such as similarity-aware instruction tuning and an evolutionary inference strategy. These components enhance POLO’s performance by providing a strong chemical foundation and amplifying the effects of the trained policy.
6. POLO’s robustness is demonstrated through ablation studies and hyperparameter analyses. The results show that POLO maintains strong performance across various settings and is particularly effective for complex, multi-objective optimization tasks.
📜Paper: arxiv.org/abs/2509.21737v1
#LeadOptimization #ReinforcementLearning #DrugDiscovery #SampleEfficiency #MolecularOptimization
1
6
856
24 Aug 2025
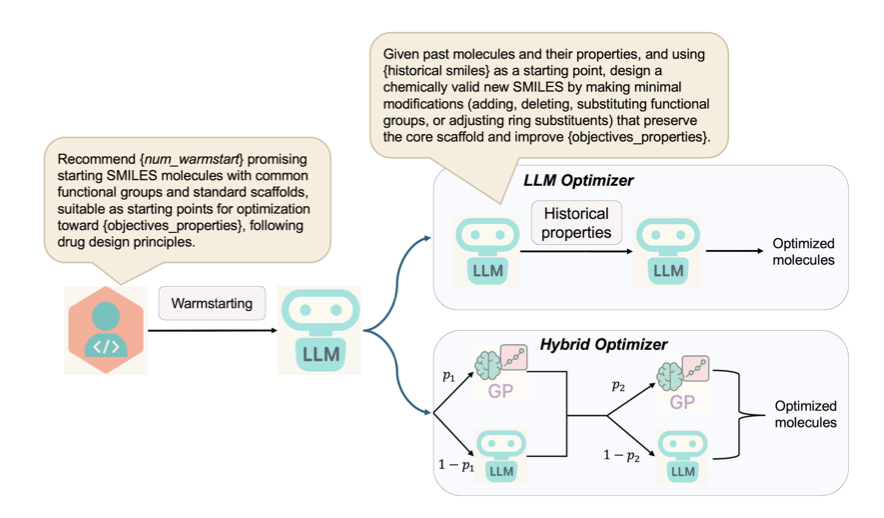
AutoLead: An LLM-Guided Bayesian Optimization Framework for Multi-Objective Lead Optimization
1. AutoLead is a groundbreaking framework that integrates Large Language Models (LLMs) with multi-objective Bayesian optimization to tackle the complex challenge of lead optimization in drug discovery. This novel approach leverages the chemical reasoning capabilities of LLMs to guide the search for novel drug-like molecules that satisfy multiple objectives, achieving state-of-the-art results on various benchmarks.
2. The framework introduces a new benchmark dataset, LipinskiFix1000, designed to evaluate lead optimization methods on realistic tasks. The goal is to modify compounds that violate Lipinski’s Rule of Five to simultaneously meet all criteria and improve their QED score. This dataset provides a rigorous testbed for multi-objective molecular design.
3. AutoLead employs a hybrid optimization strategy that dynamically alternates between LLM-driven exploration and Gaussian Process (GP)-guided exploitation. This allows for efficient navigation of the chemical space under multi-objective constraints. The LLM proposes a diverse set of candidate molecules, which are then refined by the GP surrogate model based on historical performance and uncertainty estimates.
4. The method demonstrates superior performance on multiple molecular optimization datasets, including ChatDrug-200 and DrugAssist-500, outperforming existing state-of-the-art methods in both single-objective and multi-objective tasks. The hybrid strategy (AutoLead-HO) consistently delivers top performance, especially in complex scenarios requiring simultaneous optimization of multiple properties.
5. AutoLead’s success is attributed to its ability to combine the intuitive generative power of LLMs with the uncertainty-aware search of Gaussian Processes. This integration allows for robust and scalable solutions in computational drug discovery, paving the way for more efficient and practical lead optimization pipelines.
📜Paper: biorxiv.org/content/10.1101/…
#AutoLead #DrugDiscovery #LeadOptimization #LLM #BayesianOptimization #MultiObjectiveOptimization #ComputationalBiology
1
12
1,269
8 Jun 2025
Advancing Ligand Binding Affinity Prediction with Cartesian Tensor-Based Deep Learning
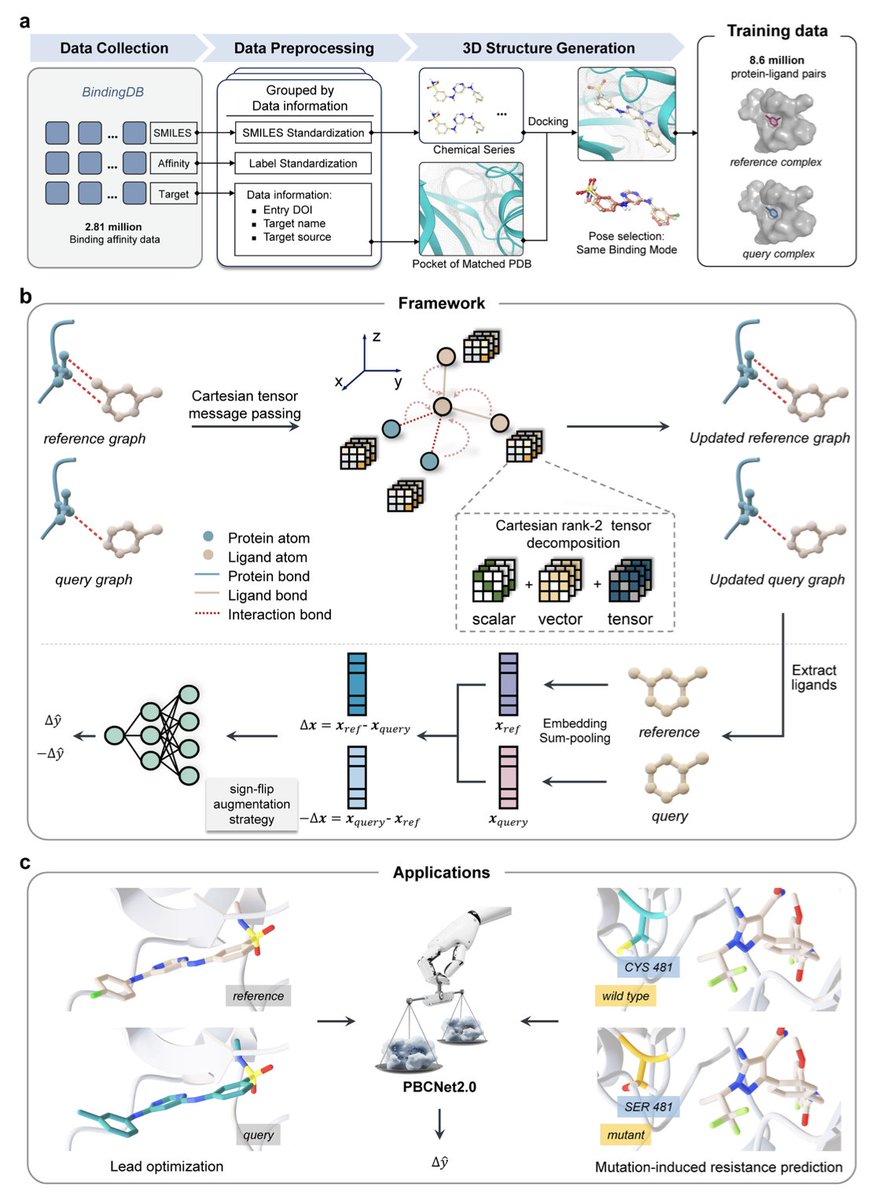
1.PBCNet2.0 is a next-generation Cartesian tensor-based Siamese Neural Network designed for protein-ligand relative binding affinity prediction, trained on an unprecedented dataset of 8.6 million complex pairs.
2.This model achieves zero-shot performance comparable to computationally intensive physics-based methods like Schrödinger’s FEP , while accelerating lead optimization by over 700% and reducing computational resource usage by 41%.
3.PBCNet2.0 leverages an equivariant graph neural network framework using Cartesian rank-2 tensors that effectively encode 3D geometric and spatial information of protein-ligand interactions without relying on predefined geometric potentials.
4.Unlike previous models, PBCNet2.0 intrinsically understands subtle molecular interactions, including fluorine orthogonal multipolar interactions with strict geometric constraints, as confirmed by interpretability analyses and benchmark tests.
5.Remarkably, PBCNet2.0 exhibits an emergent ability to predict binding affinity changes caused by protein pocket residue mutations, even though it was never explicitly trained on mutation data, highlighting potential for early identification of drug resistance mutations.
6.Retrospective and prospective experimental validations on ENPP1 and ALDH1B1 inhibitors confirm PBCNet2.0’s accuracy in predicting binding affinity differences driven by subtle molecular and stereochemical changes, supported by thermal shift assays, enzymatic inhibition, and SPR binding kinetics.
7.The model also excels at prioritizing critical binding site residues likely involved in resistance, achieving an 83% hit rate in mutagenesis validations, outperforming conventional MM-GB/SA methods.
8.While powerful, PBCNet2.0 currently relies on static protein-ligand conformations and faces challenges with negative data representing complete activity loss, pointing to future directions incorporating protein flexibility and enhanced datasets.
9.Overall, PBCNet2.0 represents a transformative, high-throughput tool that combines large-scale data, advanced geometric deep learning, and practical experimental validation to accelerate drug lead optimization and molecular probe development across diverse human protein targets.
💻Code: github.com/yujie-lab/PBCNet2
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #DeepLearning #ProteinLigandBinding #AIinPharma #LeadOptimization #StructuralBiology #MachineLearning
3
18
102
9,761
8 Jun 2025
Advancing Ligand Binding Affinity Prediction with Cartesian Tensor-Based Deep Learning
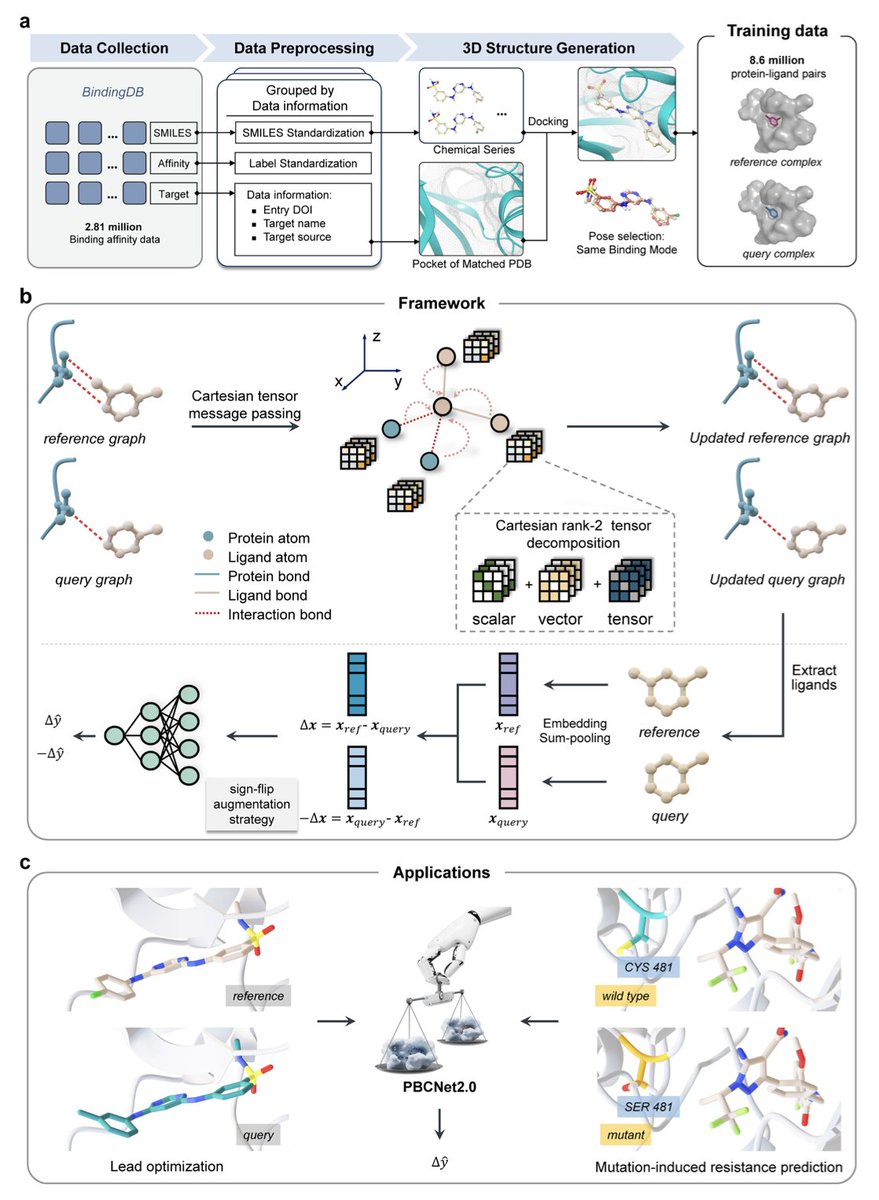
1.PBCNet2.0 is a next-generation Cartesian tensor-based Siamese Neural Network designed for protein-ligand relative binding affinity prediction, trained on an unprecedented dataset of 8.6 million complex pairs.
2.This model achieves zero-shot performance comparable to computationally intensive physics-based methods like Schrödinger’s FEP , while accelerating lead optimization by over 700% and reducing computational resource usage by 41%.
3.PBCNet2.0 leverages an equivariant graph neural network framework using Cartesian rank-2 tensors that effectively encode 3D geometric and spatial information of protein-ligand interactions without relying on predefined geometric potentials.
4.Unlike previous models, PBCNet2.0 intrinsically understands subtle molecular interactions, including fluorine orthogonal multipolar interactions with strict geometric constraints, as confirmed by interpretability analyses and benchmark tests.
5.Remarkably, PBCNet2.0 exhibits an emergent ability to predict binding affinity changes caused by protein pocket residue mutations, even though it was never explicitly trained on mutation data, highlighting potential for early identification of drug resistance mutations.
6.Retrospective and prospective experimental validations on ENPP1 and ALDH1B1 inhibitors confirm PBCNet2.0’s accuracy in predicting binding affinity differences driven by subtle molecular and stereochemical changes, supported by thermal shift assays, enzymatic inhibition, and SPR binding kinetics.
7.The model also excels at prioritizing critical binding site residues likely involved in resistance, achieving an 83% hit rate in mutagenesis validations, outperforming conventional MM-GB/SA methods.
8.While powerful, PBCNet2.0 currently relies on static protein-ligand conformations and faces challenges with negative data representing complete activity loss, pointing to future directions incorporating protein flexibility and enhanced datasets.
9.Overall, PBCNet2.0 represents a transformative, high-throughput tool that combines large-scale data, advanced geometric deep learning, and practical experimental validation to accelerate drug lead optimization and molecular probe development across diverse human protein targets.
💻Code: github.com/yujie-lab/PBCNet2
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #DeepLearning #ProteinLigandBinding #AIinPharma #LeadOptimization #StructuralBiology #MachineLearning
2
3
27
2,601

9 May 2025
The 𝗠𝗮𝗻𝗶𝗽𝗮𝗹-𝗦𝗰𝗵𝗿𝗼̈𝗱𝗶𝗻𝗴𝗲𝗿 𝗖𝗲𝗻𝘁𝗲𝗿 𝗳𝗼𝗿 𝗠𝗼𝗹𝗲𝗰𝘂𝗹𝗮𝗿 𝗦𝗶𝗺𝘂𝗹𝗮𝘁𝗶𝗼𝗻𝘀, Manipal College of Pharmaceutical Sciences (MCOPS), MAHE, Manipal, successfully conducted a one-day advanced training program on “𝘈𝘥𝘷𝘢𝘯𝘤𝘦𝘥 𝘔𝘰𝘭𝘦𝘤𝘶𝘭𝘢𝘳 𝘚𝘪𝘮𝘶𝘭𝘢𝘵𝘪𝘰𝘯𝘴 & 𝘈𝘯𝘢𝘭𝘺𝘴𝘪𝘴” on April 24, 2025.
The session brought together around 40 participants from multidisciplinary scientific backgrounds, enhancing their skills in molecular modeling, drug discovery, materials science, and computational chemistry.
Key topics covered included docking results analysis, MMGBSA, PCA, Free Energy Landscapes (FEL), WaterMap, FEP , Induced Fit Docking (IFD & IFD-MD), Binding Site Metadynamics, and de novo design of lead compounds. The program featured live Q&A sessions and one-on-one mentoring with Schrödinger experts, offering hands-on insights and personalized guidance.
This interactive, expert-led initiative fostered innovation and collaboration, equipping researchers with powerful computational tools to drive progress across pharmaceutical and chemical sciences.
#TrainingDay #MolecularSimulations #DrugDiscovery #ComputationalChemistry #Schrodinger #MolecularModeling #MaterialsScience #PharmaResearch #ScientificInnovation #MolecularDesign #LeadOptimization #DeNovoDesign #Bioinformatics #ManipalResearch #PharmaceuticalChemistry #MCOPS #MAHE #Manipal @schrodinger

1
2
152
1 May 2025
A 3D Pocket-Aware and Affinity-Guided Diffusion Model for Lead Optimization
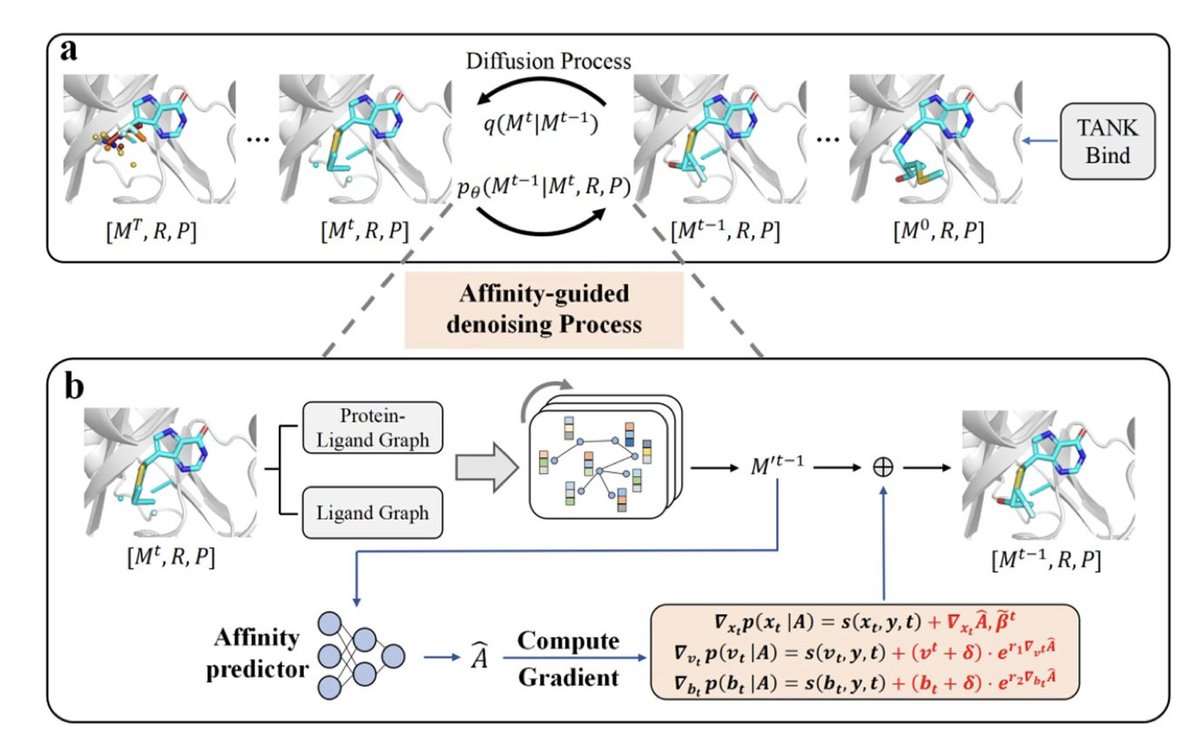
1. Diffleop introduces a novel 3D generative diffusion model that explicitly incorporates binding affinity gradients into the denoising process, guiding the generation of ligands with optimized protein binding in the context of lead optimization.
2. Unlike prior 3D pocket-aware models that only use structural features, Diffleop integrates both protein-ligand geometry and predicted binding affinity via an E(3)-equivariant graph neural network, enhancing both precision and drug-likeness of generated compounds.
3. The model operates on both continuous (atomic coordinates) and discrete (atom and bond types) noise distributions and introduces a bond diffusion mechanism to eliminate post hoc bond inference, significantly improving molecular realism.
4. A key innovation is affinity-guided denoising: at each time step, the model computes gradients of predicted binding affinity w.r.t. atom positions, atom types, and bond types to guide molecular assembly toward higher-affinity structures.
5. Diffleop significantly outperforms baseline models including GraphBP, AR, Pocket2Mol, and DiffLinker on metrics such as binding affinity (6.71 vs. 5.47–6.14) and high-affinity success rate (52.6% vs. 17.8–28.9%) in scaffold decoration and linker design tasks.
6. Beyond affinity, molecules generated by Diffleop maintain competitive scores in drug-likeness (QED), synthetic accessibility (SA), LogP, and Lipinski’s rule adherence—highlighting the model’s practicality for downstream medicinal chemistry.
7. Ablation studies reveal that removing affinity guidance reduces performance markedly (e.g., high-affinity rate drops from 52.6% to 40.4%), and excluding bond diffusion also harms chemical validity, confirming the necessity of both components.
8. The model uses equivariant graph neural networks (EGNNs) for both ligand generation and affinity prediction, and a dual-graph architecture captures both intra-ligand and protein-ligand interactions via directional message passing.
9. Unlike autoregressive models that place atoms sequentially and may introduce errors in long-range context, Diffleop uses simultaneous generation of all atoms and bonds in the masked region, resulting in more globally coherent structures.
10. Diffleop represents a next-generation framework for structure-based drug design, leveraging diffusion modeling, geometric deep learning, and knowledge-informed guidance to generate chemically valid, high-affinity molecules in 3D protein contexts.
📜Paper: arxiv.org/abs/2504.21065
#DrugDesign #MolecularGeneration #DiffusionModel #ProteinLigand #EquivariantGNN #LeadOptimization #ComputationalBiology #3DGenAI #AI4Science #StructureBasedDesign
2
19
1,374
1 May 2025
A 3D Pocket-Aware and Affinity-Guided Diffusion Model for Lead Optimization
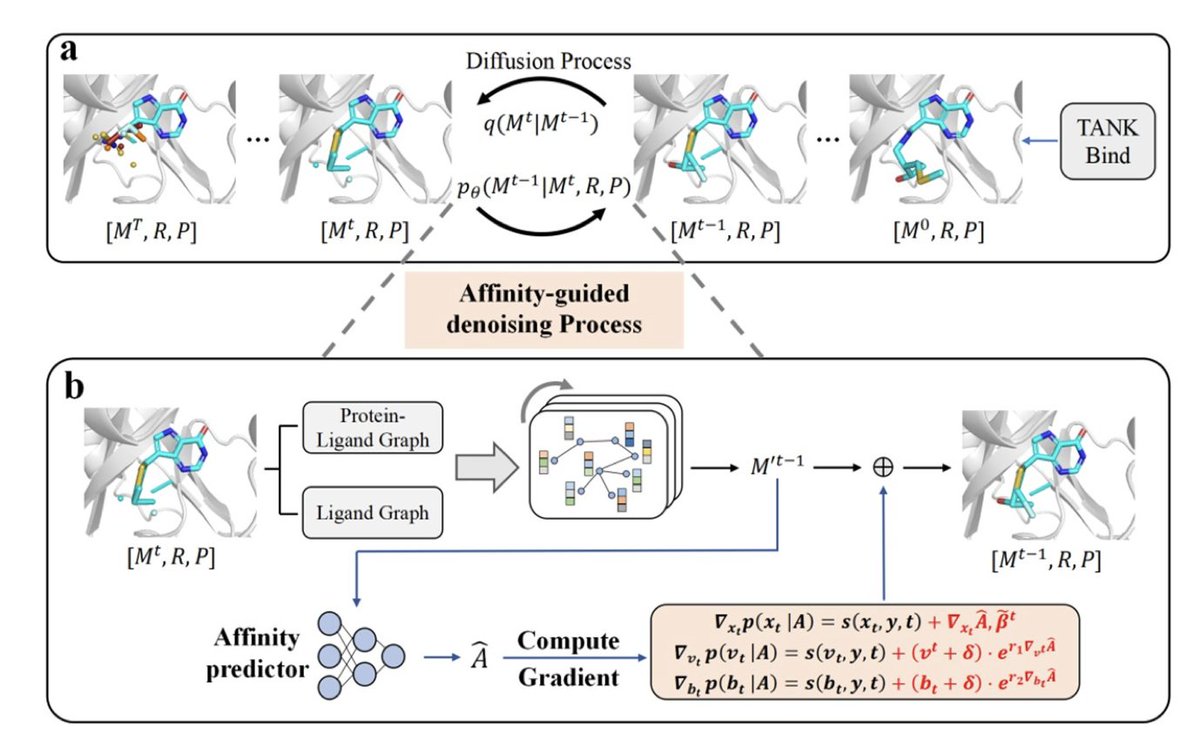
1. Diffleop introduces a novel 3D generative diffusion model that explicitly incorporates binding affinity gradients into the denoising process, guiding the generation of ligands with optimized protein binding in the context of lead optimization.
2. Unlike prior 3D pocket-aware models that only use structural features, Diffleop integrates both protein-ligand geometry and predicted binding affinity via an E(3)-equivariant graph neural network, enhancing both precision and drug-likeness of generated compounds.
3. The model operates on both continuous (atomic coordinates) and discrete (atom and bond types) noise distributions and introduces a bond diffusion mechanism to eliminate post hoc bond inference, significantly improving molecular realism.
4. A key innovation is affinity-guided denoising: at each time step, the model computes gradients of predicted binding affinity w.r.t. atom positions, atom types, and bond types to guide molecular assembly toward higher-affinity structures.
5. Diffleop significantly outperforms baseline models including GraphBP, AR, Pocket2Mol, and DiffLinker on metrics such as binding affinity (6.71 vs. 5.47–6.14) and high-affinity success rate (52.6% vs. 17.8–28.9%) in scaffold decoration and linker design tasks.
6. Beyond affinity, molecules generated by Diffleop maintain competitive scores in drug-likeness (QED), synthetic accessibility (SA), LogP, and Lipinski’s rule adherence—highlighting the model’s practicality for downstream medicinal chemistry.
7. Ablation studies reveal that removing affinity guidance reduces performance markedly (e.g., high-affinity rate drops from 52.6% to 40.4%), and excluding bond diffusion also harms chemical validity, confirming the necessity of both components.
8. The model uses equivariant graph neural networks (EGNNs) for both ligand generation and affinity prediction, and a dual-graph architecture captures both intra-ligand and protein-ligand interactions via directional message passing.
9. Unlike autoregressive models that place atoms sequentially and may introduce errors in long-range context, Diffleop uses simultaneous generation of all atoms and bonds in the masked region, resulting in more globally coherent structures.
10. Diffleop represents a next-generation framework for structure-based drug design, leveraging diffusion modeling, geometric deep learning, and knowledge-informed guidance to generate chemically valid, high-affinity molecules in 3D protein contexts.
📜Paper: arxiv.org/abs/2504.21065
#DrugDesign #MolecularGeneration #DiffusionModel #ProteinLigand #EquivariantGNN #LeadOptimization #ComputationalBiology #3DGenAI #AI4Science #StructureBasedDesign
2
29
1,663
28 Apr 2025
Improving ADME Prediction with Multitask Graph Neural Networks and Assessing Explainability in Lead Optimization
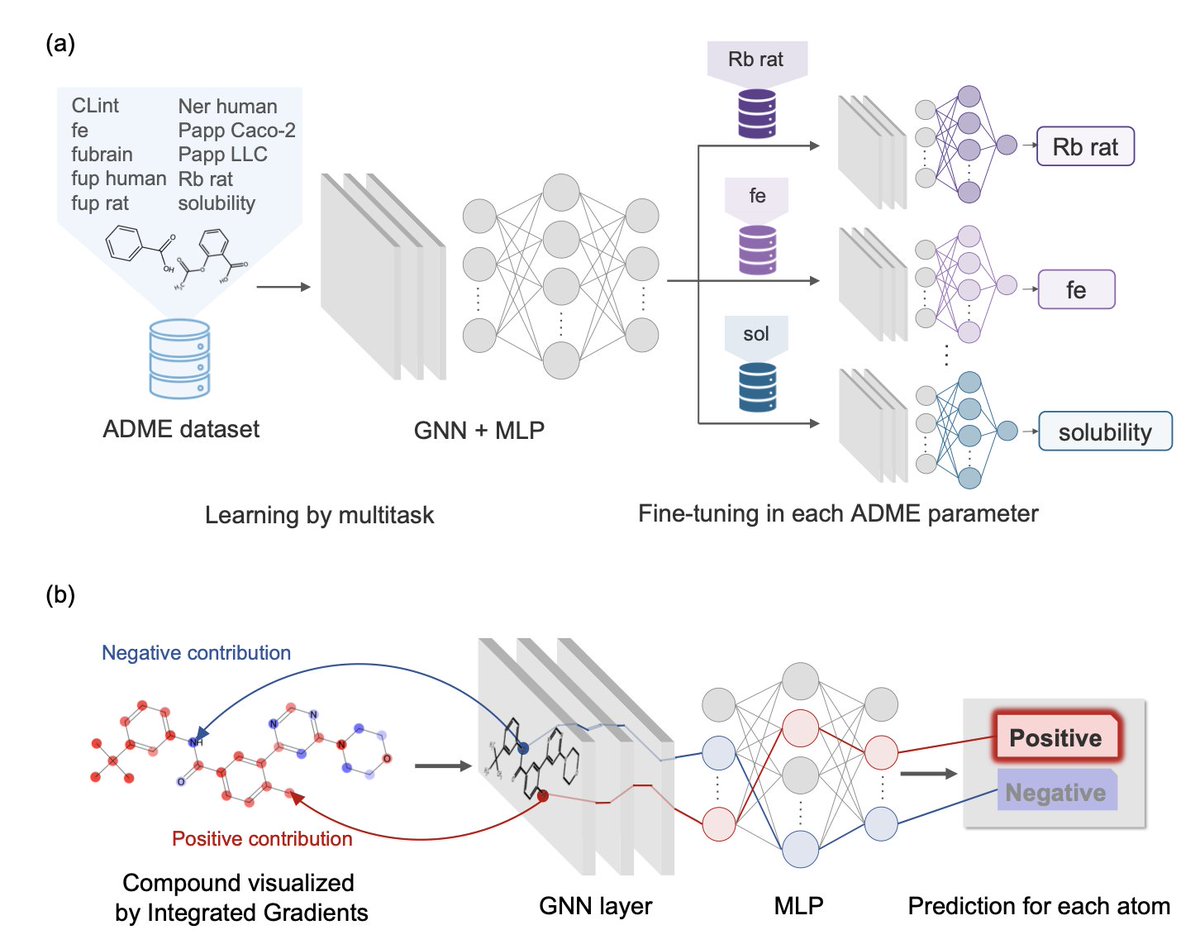
1. This study introduces a multitask graph neural network (GNNMT FT) to predict ten key ADME parameters, significantly improving early-stage drug development by addressing both data scarcity and the lack of explainability in ADME property prediction.
2. The GNNMT FT model combines multitask learning (sharing knowledge across tasks) with fine-tuning for each specific ADME property, outperforming traditional machine learning methods like random forests and single-task GNNs on seven out of ten ADME parameters.
3. Notably, GNNMT FT shows strong performance even with limited data for challenging ADME tasks such as the fraction of unbound drug in brain tissue (fubrain) and rat plasma (fup rat), achieving R² values of 0.568 and 0.655, respectively.
4. To bridge the gap between predictions and chemical insights, the authors apply Integrated Gradients (IG), a feature attribution method, to identify which molecular substructures influence ADME predictions, making the model interpretable for medicinal chemists.
5. The explainability framework was validated on real lead optimization cases from literature. For example, modifications to substructures highlighted by IG in compounds like LXH254 and GLPG-1205 aligned well with known strategies for improving metabolic stability (CLint) and permeability (Papp Caco-2).
6. The model successfully pinpointed unfavorable substructures—such as methoxy groups prone to demethylation or biaryl systems reducing solubility—providing actionable insights for optimizing compounds, demonstrated with BMS-986278 and others.
7. By integrating high-performance prediction and explainability, the GNNMT FT framework enhances lead optimization efficiency, guiding molecular design not only with accurate ADME predictions but also with interpretable rationales for structural modifications.
8. This approach shows promise in overcoming empirical limitations in ADME-driven drug design, suggesting that data-driven models with built-in interpretability could accelerate drug discovery and reduce reliance on costly in vitro/in vivo assays.
💻Code: github.com/elix-tech/kmol
📜Paper: chemrxiv.org/engage/chemrxiv…
#DrugDiscovery #ADME #MachineLearning #GraphNeuralNetworks #ExplainableAI #LeadOptimization #ComputationalBiology #Cheminformatics
6
33
1,824

7 Apr 2025
🚀 Introducing Our New FEP Service!
We are excited to introduce Free Energy Perturbation (FEP) calculations within our CADD services. It is a powerful, physics-based method that delivers highly accurate predictions of protein–ligand binding affinities, often approaching experimental-level precision.
FEP is especially valuable for:
✅ Prioritizing lead compounds with greater confidence
✅ Refining SAR with atomic-level insights
✅ Reducing the number of compounds synthesized and tested
👉 Learn more: enamine.net/services/compute…
#FEP #CADD #DrugDiscovery #LeadOptimization #ComputationalChemistry
1
5
711

24 Nov 2024
A leads tracker can help you identify which strategies are working best
#SalesStrategy #LeadOptimization #PipelineTracking #Etsy #EtsyFind
etsy.com/listing/1801319895/…
2
16
6 Nov 2024
Deep Lead Optimization: Leveraging Generative AI for Structural Modification
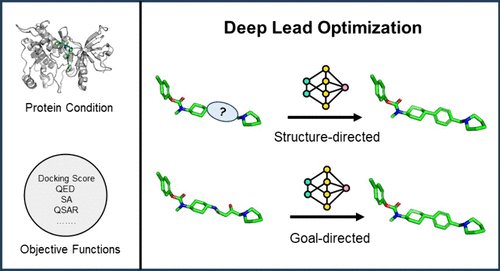
1. This paper provides a comprehensive taxonomy of generative AI-based lead optimization models, categorizing them into goal-directed and structure-directed approaches, emphasizing their unique applications and methodologies in refining molecular structures.
2. The authors highlight four key tasks in structure-directed optimization: fragment replacement, linker design, scaffold hopping, and side-chain decoration, addressing each task’s motivations, training data, and recent advancements.
3. They introduce a reference protocol for chemists to apply Generative AI in structural modification, bridging advanced computational techniques with practical drug design.
4. Unlike goal-directed methods that optimize specific properties, structure-directed models maintain specific substructures, allowing for greater control in maintaining bioactivity and therapeutic relevance.
5. Through this framework, the authors aim to advance lead optimization methods to generate compounds that better align with medicinal chemistry practices, potentially reducing drug development costs and timelines.
📜Paper: pubs.acs.org/doi/10.1021/jac…
#GenerativeAI #DrugDiscovery #LeadOptimization #MolecularDesign #ComputationalChemistry
1
16
1,577