
🇲🇾Postdoc WEHI @FelthamLab PhD @BrownLabUNSW #ubiquitin #inflammation #cholesterol 🆕️ 2024 account, past account was deleted - keen to reconnect
Joined June 2024
- Tweets 185
- Following 177
- Followers 64
- Likes 268
9 Photos and videos









jake retweeted

Mar 24
Now online! The E3-ome gene-centric compendium reveals the human E3 ligase landscape dlvr.it/TRgShy
10
33
4,127

Apr 5
Unfortunately due to word constraints and also time limitations, we didn't delve into the ligandability space and cut it out
However hoping this officially standardizes the academic and biotech landscapes - if we're all on the same page it helps propels discoveries 🙂
Mar 24

New in @CellCellPress describing the E3-ome, a compendium mapping the entire human E3 ubiquitin ligase landscape. Nice resource for targeted protein degradation research. cell.com/cell/fulltext/S0092…
28
jake retweeted

Mar 24
New in @CellCellPress describing the E3-ome, a compendium mapping the entire human E3 ubiquitin ligase landscape. Nice resource for targeted protein degradation research. cell.com/cell/fulltext/S0092…
2
11
725
jake retweeted
Now out in the April 2 issue of Cell- an important resource for the cell biology community.
The E3-ome gene-centric compendium reveals the human E3 ligase landscape: Cell cell.com/cell/fulltext/S0092…
2
3
124
jake retweeted

Apr 3
Resource @CellCellPress @WEHI_research
@mediocre_jake @BekkyFeltham
The E3-ome gene-centric compendium reveals the human E3 ligase landscape
cell.com/cell/fulltext/S0092…

2
6
627
jake retweeted

WEHI researchers have led a global study to create the first authoritative atlas for one of life’s most important enzyme families: E3 ligases.
Study led by Dr Ngee Kiat ‘Jake’ Chua (@mediocre_jake) & Dr Rebecca Feltham (@BekkyFeltham) in @CellPressNews
wehi.edu.au/news/new-enzyme-…

ALT Researchers standing in front of whiteboard with scientific information
5
22
1,432
jake retweeted

Mar 20
Great to see this work led by Bekky Feltham @WEHI_research and the global ubiquitin community now published. Happy to have contributed to the curation of HECT E3s.@CCB_Research
The E3-ome gene-centric compendium reveals the human E3 ligase landsca... sciencedirect.com/science/ar…
3
5
257
jake retweeted

ヒトのユビキチンE3リガーゼのレパートリーを定義し、ユビキチンとユビキチン様修飾系の多様性を網羅的に整理した「E3-ome」を構築し、断片化していた知見を集約することで、ユビキチンシグナル研究の基盤を提供し、新規発見の加速に寄与する論文がCell誌に発表されました。
cell.com/cell/fulltext/S0092…
3
18
1,224
jake retweeted

New Review in Trends in Cell Biology with @n_szulc
First integrative catalogue of 267 cullin-RING substrate receptors (93 linked to germline disorders) programmatic-friendly Excel resource for the community. cell.com/trends/cell-biology…
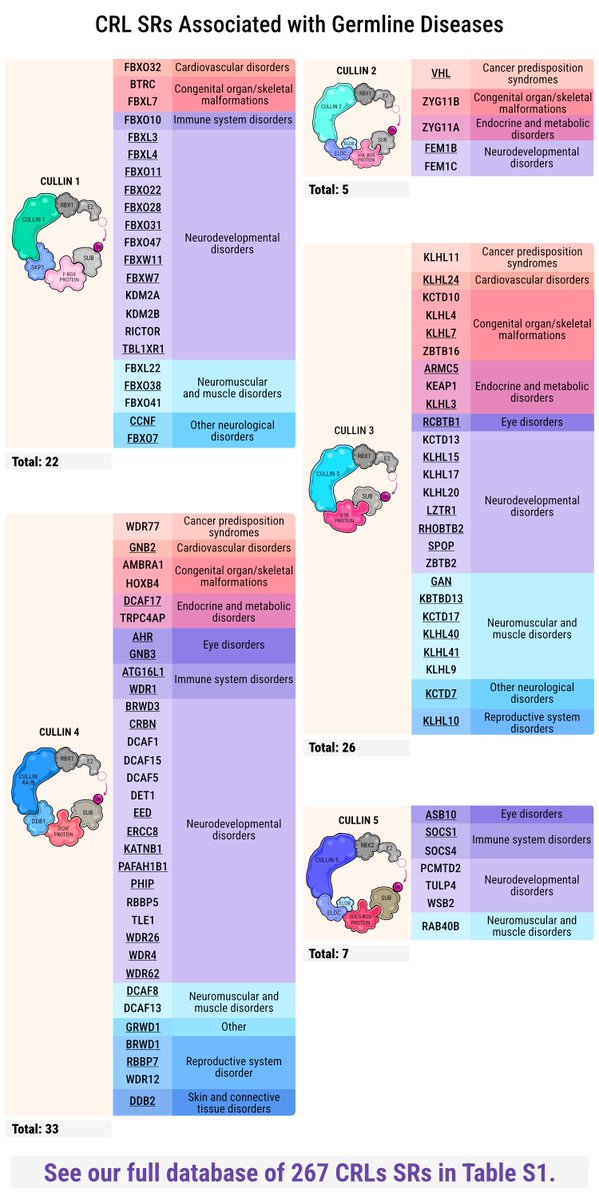
6
15
693
jake retweeted

13 Dec 2025
Wonderful joint lab holiday party with @DanNomura @RapeLab @RobertoZoncu and Saxton labs! I treasure these friendships and the community at @UCBerkeley. Happy holidays everyone! 🤗🎉




5
69
2,741
jake retweeted

14 Nov 2025
Cholesterol metabolism regulates macrophage function and inflammation-related diseases dlvr.it/TPGYpv
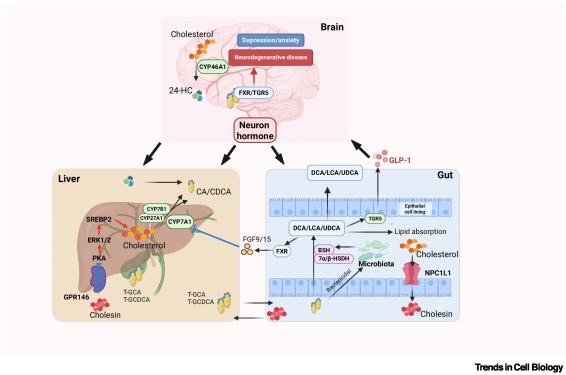
50
145
8,939
jake retweeted

12 Nov 2025
An exome-wide association study of ADHD in 8,895 cases and 53,780 controls from the iPSYCH cohort published in Nature!
Congrats to my former colleagues Jinjie Duan, Ditte Demontis, Anders Børglum and others.
Demontis et al. Nature 2025
nature.com/articles/s41586-0…
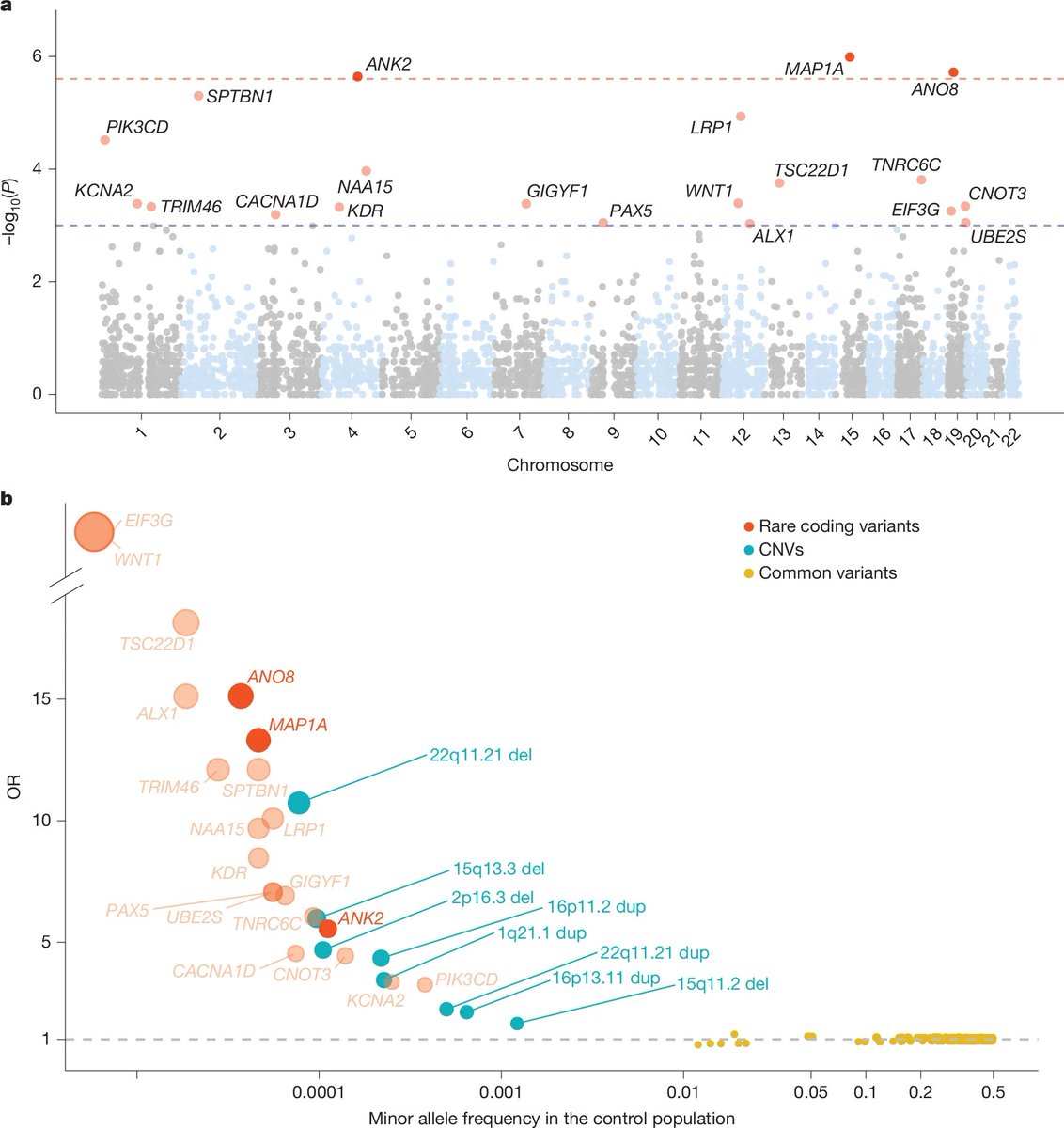
3
28
147
21,028
jake retweeted

14 Nov 2025
Postdoc time is often harder than PhD.
You have little time. Competition is higher. And you MUST stand out.
Your contract may end any moment.
Your salary is often low (for your age/education).
You’ve just relocated and prepare to relocate again.
No wonder that Max Planck Society survey showed that depression and anxiety of postdocs is rising.
Interestingly, Max Planck Institutes have substantial internal funding. So, in theory there should be less pressure to apply for grants, and everyone should be enjoying science. And yet the pressure on postdocs is immense.
📍 Many postdocs want to stay in academia.
They want to apply for faculty positions or become senior scientists.
Some of them achieve it. Others don’t.
❗️Why things don’t work out:
In a recent workshop Q&A session, I discussed various scenarios and behind-the-scenes processes that PhD students should keep in mind if they’re considering a postdoc:
“Postdocs: Competition, Hardship and Faculty Positions”
Watch it on here: youtu.be/YJm9pyb5xwY
(I’ll appreciate if you ‘like’ this video - you will GREATLY help it reach more students.)

38
253
1,278
224,982
jake retweeted
12 Nov 2025
Now online! Molecular grammars of predicted intrinsically disordered regions that span the human proteome dlvr.it/TPDd2y
27
96
15,178
jake retweeted

13 Nov 2025
Tomorrow, we have the privilege to welcome Dr. Aviv Regev @Genentech, and learn about
"From #CellAtlases to Medicines with #AI"
11/14 (Fri) 12:30 pm @ Neuroscience Research Building Auditorium, @UCLA
About the seminar series:
x.com/yk_tani/status/1970656…

ALT Dr. Aviv Regev @ Genentech on From Cell Atlases to Medicines with AI. 11/14 (Thr) 12:30 pm @ Neuroscience Research Building Auditorium, UCLA
24 Sep 2025

I am excited to welcome outstanding speakers to our
@UCLA #Bioengineering @BioEngUCLA Departmental seminar series, Fall 2025!
Please come to ENGR V 2101 at noon on Thursdays if you are around.
Link to the seminar info is in the 🧵

2
9
1,945
jake retweeted

A natural compound revitalizes the aging human immune system
nature.com/articles/s43587-0…
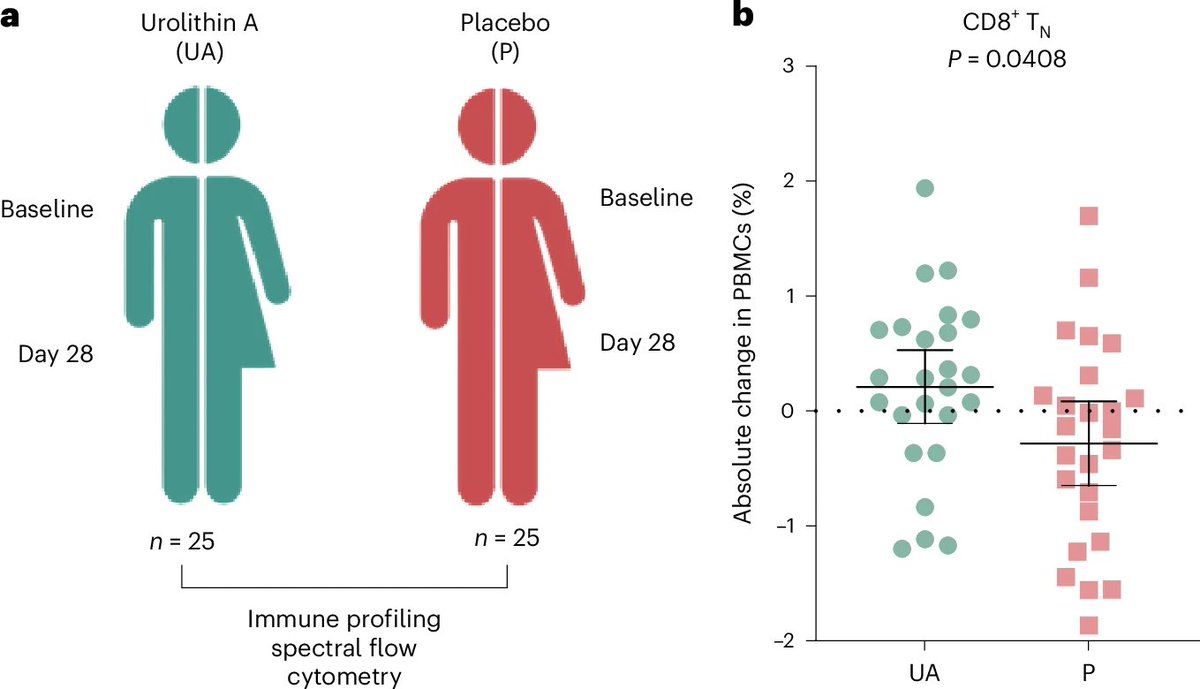
1
41
242
28,771
jake retweeted

12 Nov 2025
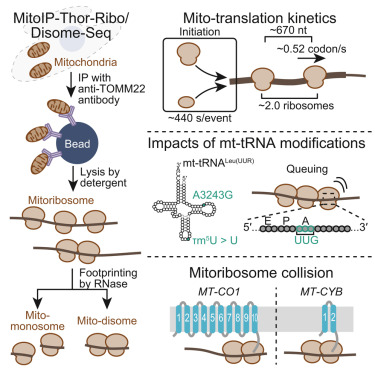
Online Now: Monitoring the complexity and dynamics of mitochondrial translation dlvr.it/TPDWC3
44
141
12,323
jake retweeted

14 Nov 2025
Chemistry writes the story of life.
Every cell, every second, it tells it again - powered by molecular design.
At the core of this process lies the citric acid cycle, the central hub of metabolism.
Here, acetyl-CoA derived from carbohydrates, fats, or proteins enters a precise series of reactions that convert fuel into energy.
Each step transfers electrons, drives ATP synthesis, and sustains the continuous renewal of life at the cellular level.
What may seem invisible is, in fact, the most constant motion in existence; the quiet rhythm of biochemistry that powers everything we do.
22
366
1,291
52,944
jake retweeted

24 Oct 2025
Predicting protein-protein interactions in the human proteome
Predicting which human proteins shake hands—and how—is a longstanding bottleneck. Proteins rarely act alone; they assemble into complexes that drive immunity, metabolism, signaling, and disease. But testing hundreds of millions of possible pairs experimentally is slow, expensive, and blind to many weak or transient interactions.
Jing Zhang, Qian Cong, David Baker and coauthors tackle this with a smart AI data pipeline. First, they amplify evolutionary “clues” by assembling omicMSAs—deep multiple sequence alignments mined from petabytes of raw eukaryotic genomic data—so coevolution across species pops out. Second, they train a fast interaction model, RoseTTAFold2-PPI, not just on scarce complex structures, but on domain–domain contacts distilled from ~200M AlphaFold monomers—a huge synthetic training set that teaches the network what real interfaces look like.
The payoff is big: a proteome-scale screen over ~200M human pairs yields ~18,000 PPIs at ~90% precision (and ~29k at 80%), including ~3,600 not previously reported. The method excels on transmembrane interactions, a class that’s notoriously hard in the lab, and produces 3D complex models—so you don’t just get a yes/no, you see the interface. Mapping human variants onto these models flags ~4,950 PPIs with disease mutations at the contact surface, offering concrete hypotheses for mechanism.
Beyond pairs, the team reconstructs higher-order assemblies and nominates new components for well-studied complexes (e.g., telomere maintenance, GPI-GnT, cilia/flagella machinery), and highlights GPCR partners and mitochondrial modules that have been hiding in plain sight.
Stepping back: this is a credible path toward a computed 3D human interactome—faster, cheaper, and increasingly comprehensive as more genomes and structures arrive. It doesn’t replace experiments; it prioritizes them, focusing bench time where the biology is richest.
Paper: science.org/doi/full/10.1126…

2
88
428
29,111
jake retweeted

23 Oct 2025
A thoughtful highlight of our results on alternate RNA decoding:
Highlight: science.org/content/blog-pos…
Preprint: biorxiv.org/content/10.1101/…

8
31
2,895