
Scientist. Personal account.
Joined September 2022
- Tweets 1,280
- Following 167
- Followers 296
- Likes 1,576
17 Photos and videos









Pinned Tweet

Feb 12
Our latest manuscript is finally out in @NatureSMB!
Here, we show that the GAP activity of SEAC (GATOR) acts as a molecular switch, regulating both inhibition and reactivation of TORC1 upon changes in amino acid levels.
nature.com/articles/s41594-0…
2
8
25
2,383
Lucas Tafur retweeted

Jun 10
Always good to remember that the PDB sequence of a structure is not always the sequence of the protein made nor the sequence of the protein analyzed, its the sequence of the protein a human remembered to type in.
3
5
70
9,830
Lucas Tafur retweeted

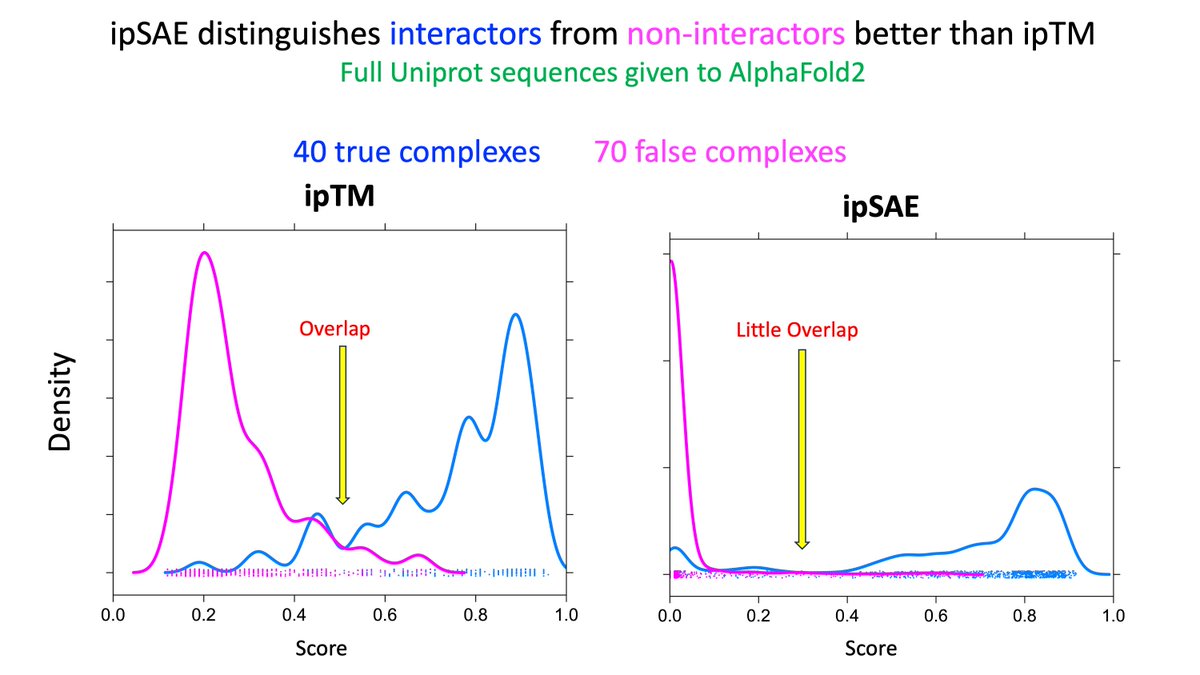
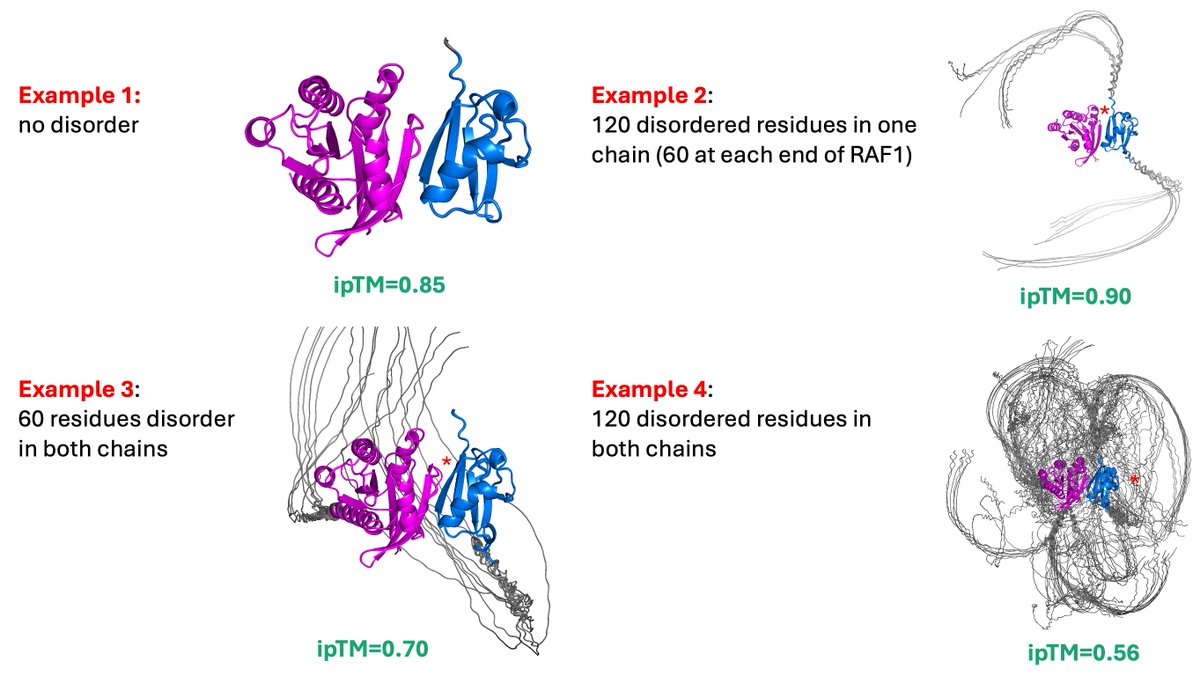
STOP using ipTM for AlphaFold PPI screens.
* Artificially LOW when both full-length proteins have irrelevant, non-binding IDRs or domains.
* If one protein is folded & the other contains irrelevant IDRs or domains, the score is too HIGH because of d0.
* Use ipSAE or LIS.

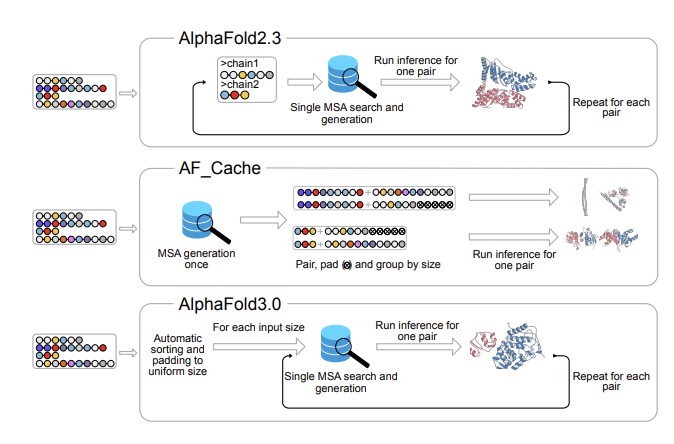
AF Cache: Efficient Pipeline for Running AlphaFold for High-Throughput Protein-Protein Interaction Prediction
1 AF Cache is a Nextflow pipeline designed to make large-scale protein–protein interaction (PPI) screening practical with AlphaFold2 and AlphaFold3 by cutting the dominant runtime bottlenecks: repeated MSA generation and (for AF2) repeated JAX recompilations.
2 The key idea is to generate MSAs once per unique monomer for the whole dataset, then reuse (“cache”) them across all pairs. For an all-against-all screen of N proteins, default multimer workflows can trigger ~N(N 1) chain-level MSA generations, while AF Cache reduces this to N by construction.
3 AF Cache accelerates MSA generation by using GPU-accelerated MMseqs2 and batching all proteins in a single dataset-level step (rather than per-target or small batches). It also overlaps CPU and GPU steps across UniRef and environmental database alignments to reduce GPU idle time.
4 For AlphaFold2 specifically, AF Cache adds sequence-length bucketing for multimers (bucketed by total chain length) and pads within each bucket so JAX compilation happens once per bucket instead of once per pair—saving ~1–2 minutes of GPU time for every additional multimer in that bucket.
5 Benchmark: 100 human mitochondrial proteins (40–1000 aa) were screened all-against-all including homodimers (5,050 pairs). Settings were streamlined for throughput (AF2: single model, 3 recycles, templates disabled; AF3: single seed, one diffusion sample).
6 Pre-prediction speedups (MSA stage): when comparing to a realistic 128-CPU-core baseline, AF Cache achieved ~13x faster MSA generation for AF2 settings using full BFD, and ~5x faster for AF3 settings using small BFD. The paper also reports very large raw GPU-vs-CPU core-hour reductions, emphasizing the benefit of GPU-based MSA search in high-throughput regimes.
7 End-to-end impact for AF2: caching bucketing reduced AF2 prediction compilation time from 253 to 125 GPU hours (>50% reduction), and made inference ~90 seconds faster per protein pair on average while preserving the same runtime scaling with pair length.
8 Output similarity was assessed via ipTM comparisons. Correlations between default vs AF Cache runs were moderate across all pairs (Pearson r ~0.70 for AF2; ~0.64 for AF3), but much higher for pairs where both proteins map to a shared PDB entry (r ~0.98 for AF2; ~0.94 for AF3), suggesting structurally supported cases are highly consistent across pipelines.
9 Practical deployment details: AF Cache provides a ready-to-use workflow for local and HPC environments, supports all-against-all or user-specified pair lists, automates AF3 JSON preparation, and can download/install dependencies (including databases; AF2 network parameters) as needed.
💻Code: github.com/clami66/AF_cache
📜Paper: arxiv.org/abs/2606.04566
#AlphaFold #ProteinInteractions #PPI #Bioinformatics #ComputationalBiology #StructuralBioinformatics #Nextflow #HPC #MMseqs2 #Proteomics
5
36
211
17,930
Jun 7
Sooner, rather than later, the AI tech bro startup biotech industry will burst…

This is how you know tech bros are not going to solve major problems in biology.
1
2
4
668
Lucas Tafur retweeted

Jun 3
Exciting new work from @RobertoZoncu, led by all star first authors @clairegoul and @aakritijain24 reveals the mechanism mediating the sensing of lysosomal damage and recruitment of ESCRT for membrane repair. Beautiful work!🤩
nature.com/articles/s41586-0…
14
54
3,600
Lucas Tafur retweeted

1 Jun 2025
Just when you think this nonsense can't get any worse, you find this hilarious attempt to lie about the lie:
- we made a mistake and did not include the pre-registered primary endpoint when we full-well knew and then attempted to obscure the data and did not include it any of our social media tsunami in early April, nor was it even discussed in the paper, but in this video we admit we had it but just did not like it
- but then people went crazy so
- we are crafting a story that we were naive and really never should have made this the primary endpoint and actually we really have another way to do this that was the primary methodology (WTF) and should have been the primary endpoint
- and now we will spin on and on about how the Cleerly results are just implausibly bad and so must be wrong even though we never even discussed this for one second in our social or in the actual paper
- and also it was probably covid or "some covid-related activity" that led to these Cleerly results being so messed up
- also here are new data with another method (keep re-analysing it till it looks right) and these data look less bad so it's all good...
Seriously, i thought this was a clown show but now I am searching for something that more accurately describes it. I am not sure I can solve that
1 Jun 2025

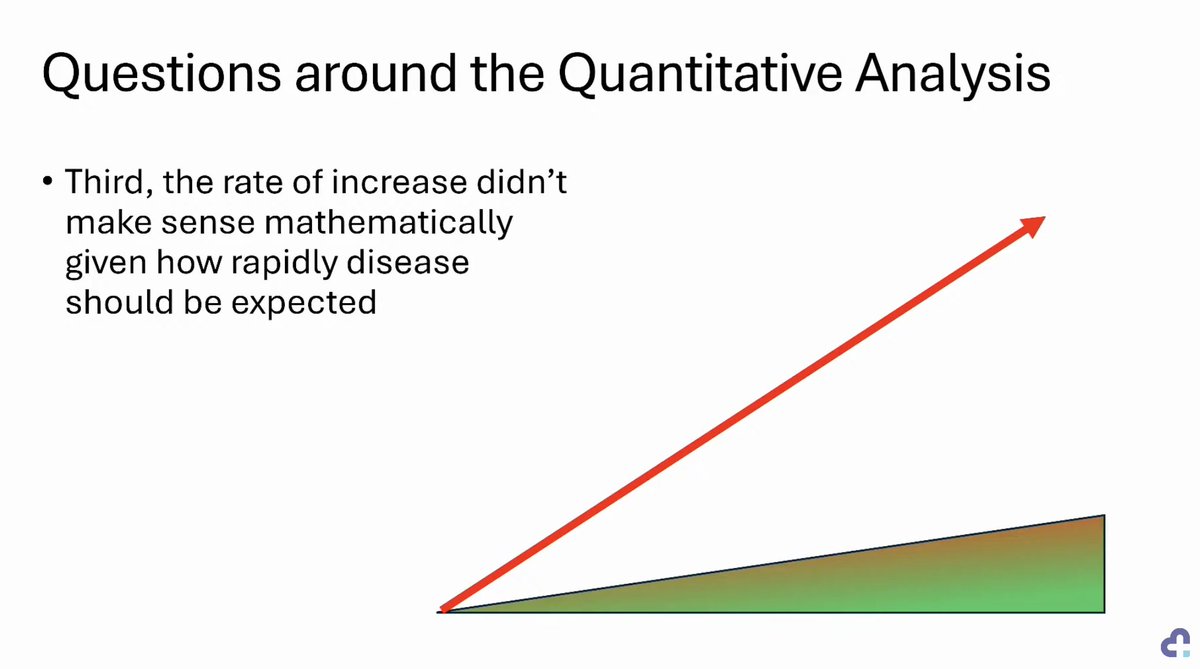
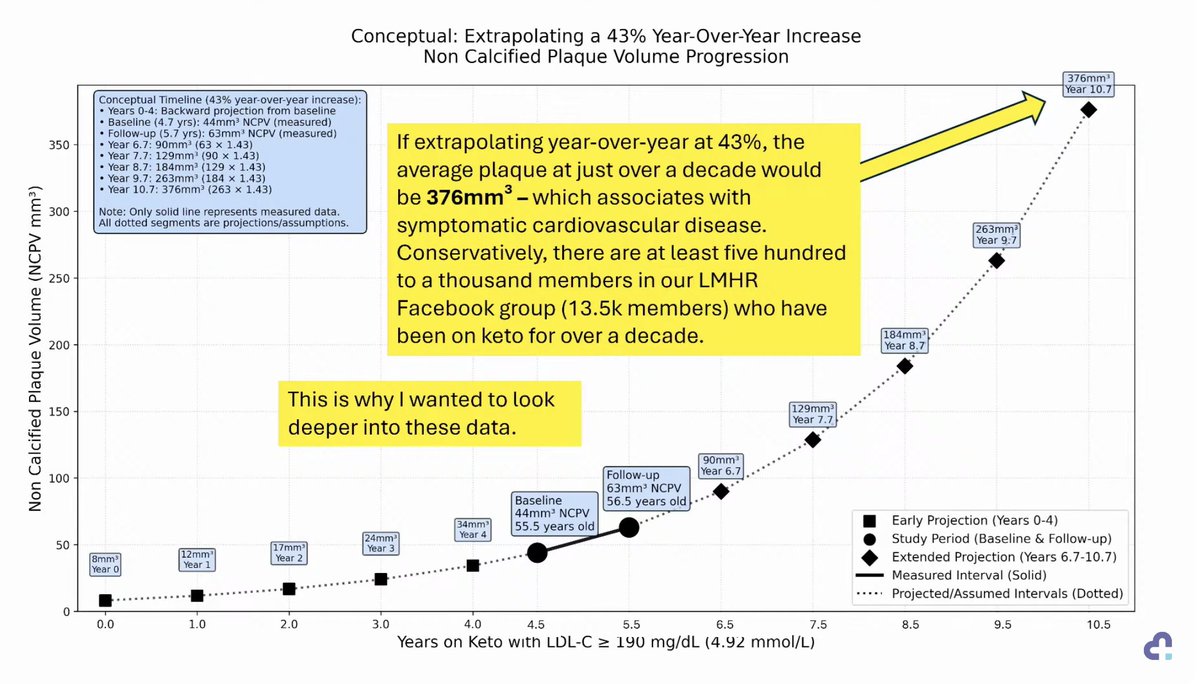
KETO-CTA: Schrödinger's Plaque 🥓
In his latest lecture, @realDaveFeldman claims plaque progression was TOO RAPID to make sense.
Yet, we were told "NO or MINIMAL plaque progression".
Can progression be so great as to not make sense AND also "no or minimal"?
14
23
115
37,215
Lucas Tafur retweeted

Jun 2
Online now!✨Iiams, Skinner et al explore the effect of early-onset, lifelong time-restricted feeding in >500 male and female mice, and report extended healthspan in male and female mice, and extended lifespan in male mice @SamIiamsClever @CircadianClocks nature.com/articles/s43587-0…
1
14
42
6,171
Lucas Tafur retweeted

Jun 1
Hello world, meet 1,000× Expansion Microscopy.
1,000,000,000× expansion by volume! A gel that starts at a few centimeters will then expand to the volume of an Olympic swimming pool. biorxiv.org/content/10.64898…
In our new bioRxiv preprint, work carried out between MIT and UMG, led by Helena Hu in collaboration with scientists from the labs of @eboyden3 Ed Boyden, Silvio Rizzoli, and myself, we present Thousandfold Expansion Microscopy.
By enlarging biological specimens across multiple rounds of expansion, molecular-scale features, as small as the distances between adjacent amino acids, can be visualized with conventional optical microscopes.
Democratizing super-resolution microscopy.

12
139
599
96,435
Lucas Tafur retweeted

The reviewers are us
May 27

Im not sure I will ever overcome how furious I get reading reviewer comments.
You spend 3 years of your life working on something just for 3 people to skim your paper and point out a few things that were already well explained in an incredibly rude way and then reject it
1
2
11
2,804
Lucas Tafur retweeted

May 27
New publication from our group: what happens inside human endometrial tumors during a ketogenic diet?
We analyzed paired tumor biopsies before and after a very low-carbohydrate intervention in women with endometrial cancer 🧵
3
19
61
13,845
Lucas Tafur retweeted
MD simulations of proteins have been around for almost 50 years (1977). What are you talking about?
May 16

For the past 70 years, modern biology has been built on a static objects worldview: DNA = information, proteins = structures, cells = nanomachines, disease = broken parts, drugs = part repair/update.
I'm honestly excited for a “new worldview” and “Dynamic Biology” to emerge.
14
23
246
40,641
May 3
👇👇 the best evidence *for you* is N=1.
Ok we are closer to agreement than it first appeared.
N=1 experiments that are not randomized are equivalent to case reports, especially if documented to a similar degree.
N=1 randomized experiments are the highest tier evidence for the individual in whom they are done provided that the time course is feasible and relevant to the outcome of interest.
N=1 randomized experiments cannot be used for population-level guidelines because they don't apply outside the individual.
However, since all information from any source is imperfect, and since many areas of interest have enormous gaps in knowledge, it is very valuable to use findings from N=1 experiments as potentially relevant to other individuals especially when those individuals are similar in genetics, history, and constitution in the variables thought to be relevant to inter-individual differences.
Randomized self-experiments are practical on short time scales, especially when effects are acute over the course of hours. They are reasonably practical for time horizons of one day or several days. They are entirely feasible for time horizons that take weeks or longer but are difficult enough at those timescales that one would have to be extremely motivated to do it, either because the stakes seemed high or because one is a scientist in the classical sort, which is now almost absent from science.
The classical scientist, the most genuine of all scientists, loved to experiment out of curiosity and loved to experiment on himself.
There are many such scientists in history but they are a rare breed.
Barry Marshall most famously with ulcers, Carl Wilhelm Scheele most notoriously for smelling and tasting every chemical he could, William Brown who working with George Burr tried to give himself an EFA deficiency.
A R Berger 1, H H Schaumburg, C Schroeder, S Apfel, and R Reynolds taught us a lot about vitamin B6 toxicity by purposefully inducing it in themselves in self-experiments that lasted YEARS. They published their report in 1992.
So the answer to your question is that no it is not practical but it is the sign of a genuine scientist of the highest breed.
75
Lucas Tafur retweeted

May 2
Wow. Someone was asleep at the editorial desk. “Reduced” as a causal verb just doesn’t belong h/t @djc795
May 2

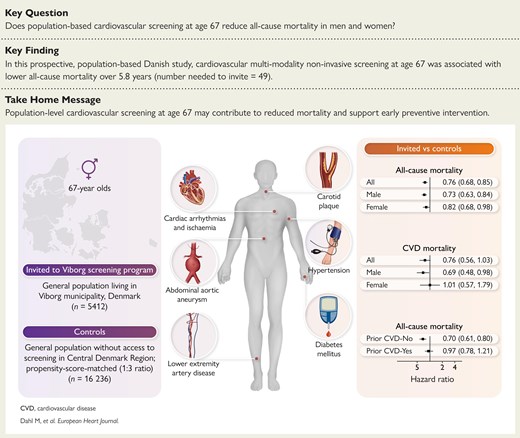
Multimodality non-invasive cardiovascular screening reduces 5-year all-cause mortality in both sexes!
doi.org/10.1093/eurheartj/eh…
#multimodality #screening #cardiotwitter @ESC_Journals @escardio
3
7
56
27,418
May 2
Beautiful

Water is 70–80% of a cell, with our mspSA SGAG affinity grid, we capture a near atomic (<2 Å) view showing water actively driving gene transcription inside RNA Pol II. Not just background,water is part of the machinery. Now published in Molecular Cell.doi.org/10.1016/j.molcel.202…
69
Lucas Tafur retweeted

Nature Human Behaviour から "How to design effective scientific figures" というコメンタリーが出ました🎉参考になれば幸いです!!
nature.com/articles/s41562-0…
@NatureHumBehav


I’m thrilled to announce that my new comment titled “How to design effective scientific figures” is now released from @NatureHumBehav
This provides five keys to make your graphical items attractive 🎨 Hope this is useful for your project!
nature.com/articles/s41562-0…
14
222
1,418
296,112
Lucas Tafur retweeted

Apr 30
We propose a two-branched structure of causal pathway of cancer cachexia, with one energy balance branch and the other independent of energy balance.
Separating food intake-dependent and -independent effects in cancer cachexia: iScience cell.com/iscience/fulltext/S…
1
6
21
1,428
Lucas Tafur retweeted

Mechanical load inhibits cancer growth in mouse and human hearts | Science science.org/doi/10.1126/scie…
11
41
2,187
Lucas Tafur retweeted

Every time I tell AI utopianists that biology is too complex for AI to "solve", they cite the success of AlphaFold.
No, AlphaFold did not "solve" protein folding. It gets broad structures correct ~70-88% of the time (depending on evaluation), enabling useful but flawed statistical guesses.
True "solving" would require ~99.9% accuracy, practically zero meaningful edge cases, and high confidence across fine details like side chains and conformations.
Even then, this is just one narrow slice of the complexities of proteomics.
The persistent gap between the "AlphaFold solved protein folding" claim and reality is a perfect example of AI overhype in biology.
89
134
837
122,247
Lucas Tafur retweeted

Apr 24
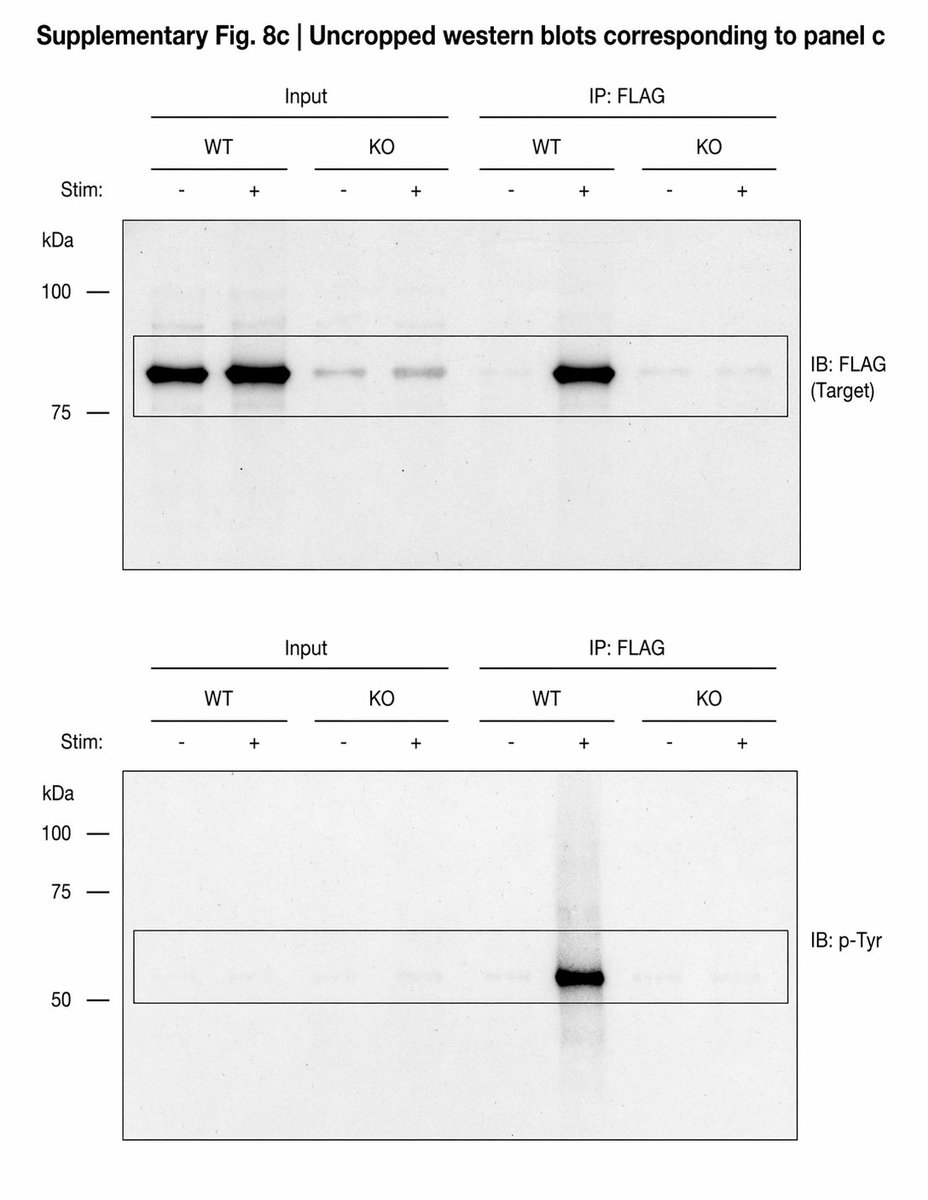
⚠️Requesting uncropped gels might not be enough!⚠️ Those are FABRICATED gels by ChatGPT Image 2.0 ❗️❗️❗️ I am not an expert but would get fooled. @Thatsregrettab1
15
16
79
16,900

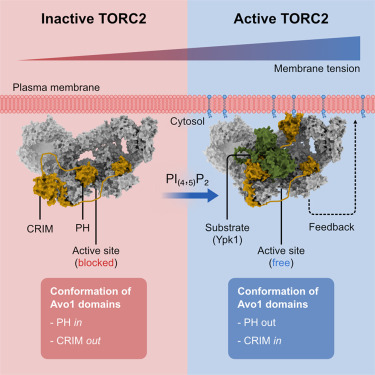
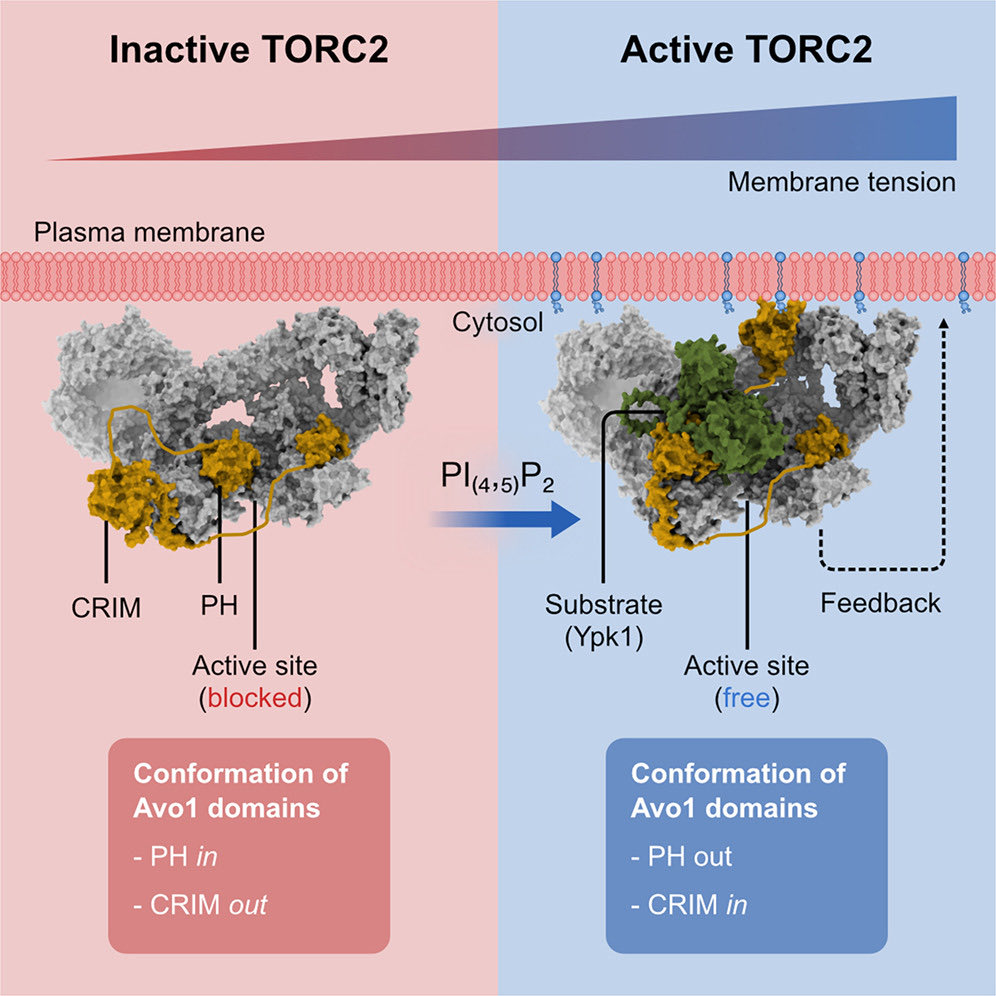
Apr 16
Happy to share our latest paper in @MolecularCell!
Here, we determined the cryo-EM structure of endogenous TORC2 to up to ~2.2Å resolution.
Combined with structure predictions and in vivo experiments, we reveal how signaling lipids regulate TORC2 activity.
1
2
6
1,181