
16 May 2025
A Comparative Review of RNA Language Models
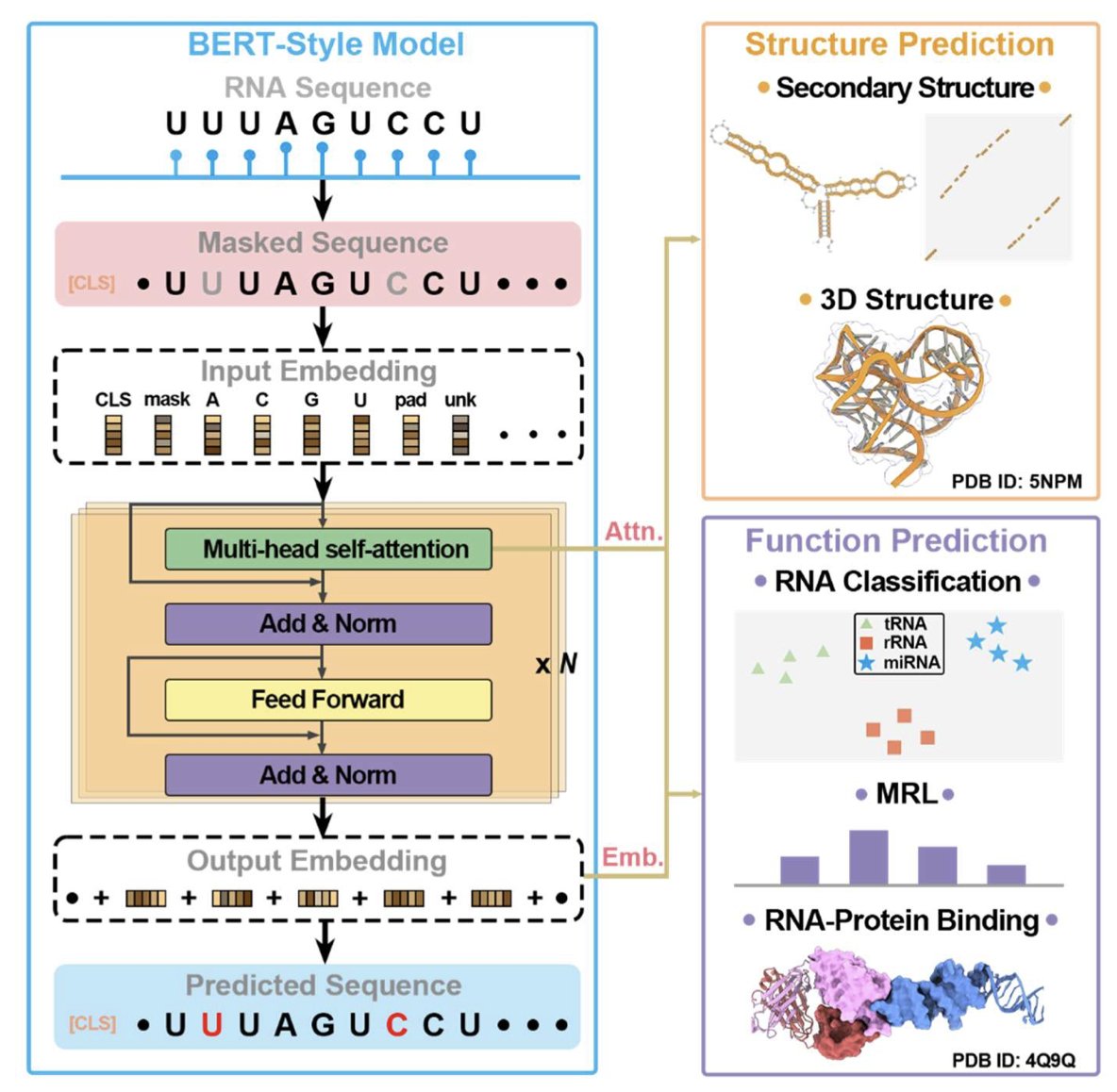
1.This review systematically benchmarks 13 RNA language models (LMs) across structural and functional tasks, revealing that models excelling in one often underperform in the other—highlighting the challenge of balanced unsupervised training for RNA.
2.RNA LMs are categorized into three classes: Class I models trained on diverse RNA types (esp. non-coding RNAs), Class II models trained on specific RNA subsets (e.g., UTRs), and Class III unified models trained on DNA, RNA, and/or proteins.
3.Zero-shot secondary structure prediction shows RNA-MSM (MSA-based) achieves the highest F1 score (0.631), outperforming all single-sequence models—even those with 10× larger parameter counts like AIDO.RNA (1.6B).
4.Interestingly, the protein LM ESM2 (15B) performs better than most RNA LMs on RNA structure prediction (F1 = 0.675), underscoring the limited training of RNA models and perhaps the benefits of massive pretraining.
5.Models like MP-RNA and RNA-km rank high on structural prediction but perform poorly on functional classification, indicating overfitting to structural features and lack of embedding generality.
6.Zero-shot classification tasks using cosine similarity between embeddings show RNA-FM achieves the lowest overlap ratio (OR = 0.070) in Rfam—demonstrating superior ability to distinguish RNA families in an unsupervised manner.
7.Only a few models, notably AIDO.RNA (1.6B), perform relatively well on both tasks—though performance gain in secondary structure came at the cost of higher OR in classification, again underscoring the tradeoff.
8.Unified LMs like LucaOne underperform Class I RNA LMs on both structure and function, suggesting that multitask pretraining across DNA, RNA, and proteins may dilute RNA-specific signal.
9.The survey also highlights key design innovations across models—such as motif-aware masking (RNAErnie), k-mer input (RNA-km), cross-modality pretraining (ProtRNA), and MSA-augmented structure learning (RNA-MSM).
10.Dataset bias is a major concern: most pretraining and benchmarking datasets are heavily skewed toward tRNA and rRNA, with many evaluations lacking proper redundancy removal—raising questions about generalization.
11.Model size alone does not ensure superior performance. While larger models (e.g., AIDO.RNA 1.6B) help structure prediction, they can degrade functional performance and do not match the structural clarity of MSA-based methods.
12.The authors emphasize the need for unbiased multi-objective training, better diversity in training datasets, and model designs that preserve both global structural constraints and fine-grained functional semantics in RNA sequences.
📜Paper: arxiv.org/abs/2505.09087
#RNA #LanguageModels #RNAstructure #RNAfunction #LLM #Bioinformatics #AI4Science #MSA #SecondaryStructure #ncRNA #ComparativeEvaluation #TransformerModels
3
16
67
6,142
16 May 2025
A Comparative Review of RNA Language Models
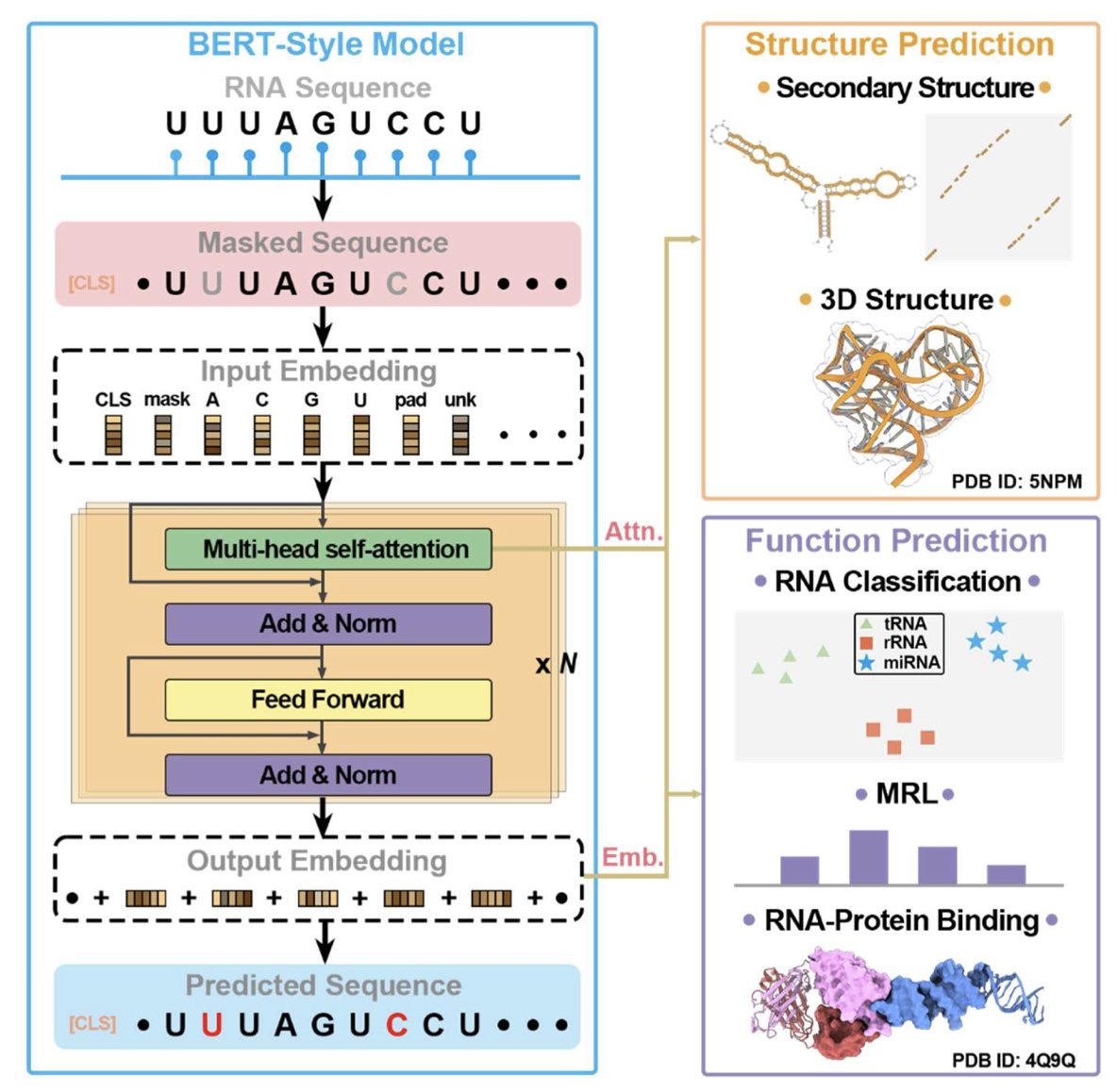
1.This review systematically benchmarks 13 RNA language models (LMs) across structural and functional tasks, revealing that models excelling in one often underperform in the other—highlighting the challenge of balanced unsupervised training for RNA.
2.RNA LMs are categorized into three classes: Class I models trained on diverse RNA types (esp. non-coding RNAs), Class II models trained on specific RNA subsets (e.g., UTRs), and Class III unified models trained on DNA, RNA, and/or proteins.
3.Zero-shot secondary structure prediction shows RNA-MSM (MSA-based) achieves the highest F1 score (0.631), outperforming all single-sequence models—even those with 10× larger parameter counts like AIDO.RNA (1.6B).
4.Interestingly, the protein LM ESM2 (15B) performs better than most RNA LMs on RNA structure prediction (F1 = 0.675), underscoring the limited training of RNA models and perhaps the benefits of massive pretraining.
5.Models like MP-RNA and RNA-km rank high on structural prediction but perform poorly on functional classification, indicating overfitting to structural features and lack of embedding generality.
6.Zero-shot classification tasks using cosine similarity between embeddings show RNA-FM achieves the lowest overlap ratio (OR = 0.070) in Rfam—demonstrating superior ability to distinguish RNA families in an unsupervised manner.
7.Only a few models, notably AIDO.RNA (1.6B), perform relatively well on both tasks—though performance gain in secondary structure came at the cost of higher OR in classification, again underscoring the tradeoff.
8.Unified LMs like LucaOne underperform Class I RNA LMs on both structure and function, suggesting that multitask pretraining across DNA, RNA, and proteins may dilute RNA-specific signal.
9.The survey also highlights key design innovations across models—such as motif-aware masking (RNAErnie), k-mer input (RNA-km), cross-modality pretraining (ProtRNA), and MSA-augmented structure learning (RNA-MSM).
10.Dataset bias is a major concern: most pretraining and benchmarking datasets are heavily skewed toward tRNA and rRNA, with many evaluations lacking proper redundancy removal—raising questions about generalization.
11.Model size alone does not ensure superior performance. While larger models (e.g., AIDO.RNA 1.6B) help structure prediction, they can degrade functional performance and do not match the structural clarity of MSA-based methods.
12.The authors emphasize the need for unbiased multi-objective training, better diversity in training datasets, and model designs that preserve both global structural constraints and fine-grained functional semantics in RNA sequences.
📜Paper: arxiv.org/abs/2505.09087
#RNA #LanguageModels #RNAstructure #RNAfunction #LLM #Bioinformatics #AI4Science #MSA #SecondaryStructure #ncRNA #ComparativeEvaluation #TransformerModels
3
11
876
2 May 2025
FlatProt: 2D visualization eases protein structure comparison
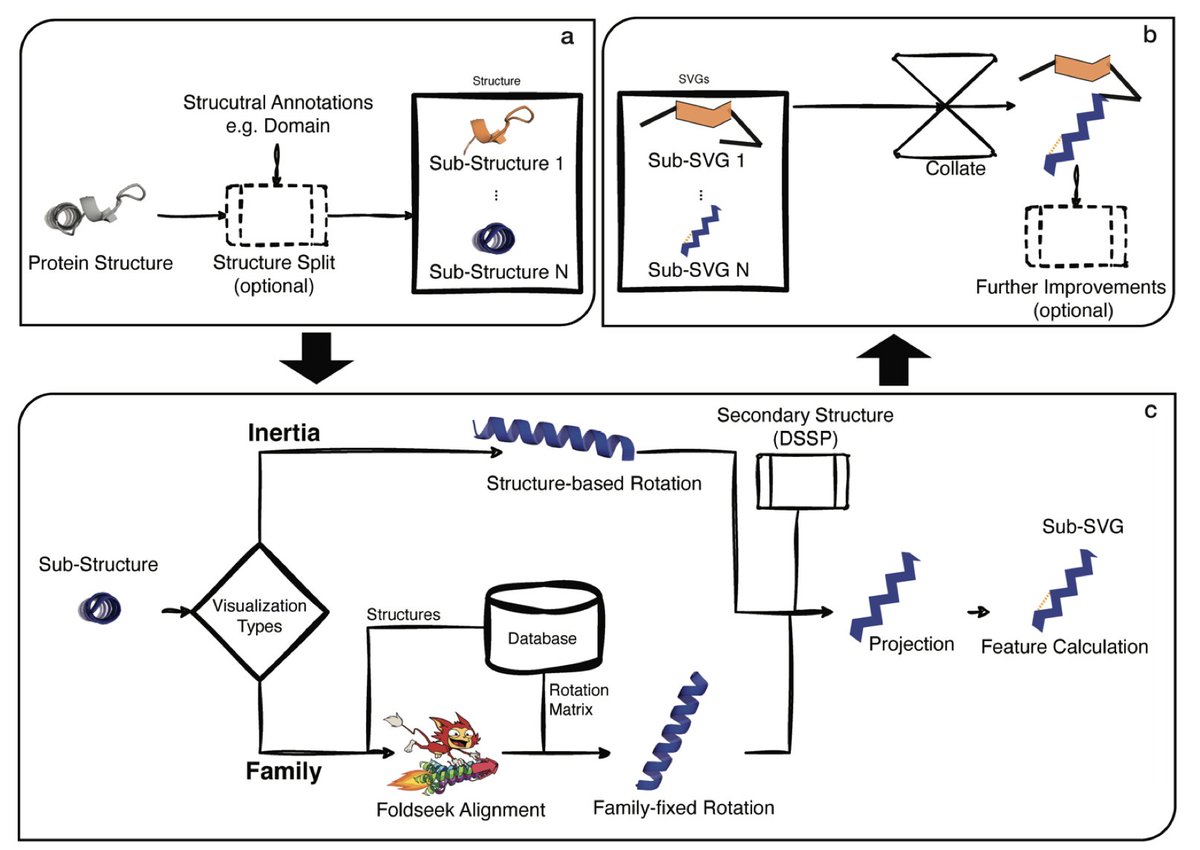
1. FlatProt is a new tool that standardizes 2D visualizations of 3D protein structures, enabling consistent, scalable, and intuitive comparison of protein families across entire proteomes.
2. The tool solves a fundamental bottleneck in structural biology: viewing and comparing many 3D protein structures on 2D screens without losing key information—an essential task in drug design and protein function analysis.
3. FlatProt employs both inertia-based and family-based structure rotation to project proteins consistently into 2D space, preserving structural orientation across related proteins and improving comparability.
4. It supports domain-aware visualizations, simplified segment representations (e.g., helices as zigzags, strands as triangles), and overlay of annotations like disulfide bonds or divergent regions via lDDT scores.
5. The tool enables consistent visualization for structurally diverse families, like Three-Finger Toxins (3FTx) and Kallikreins (KLK), making it easier to detect conserved features and family-specific variations.
6. Unlike prior tools (e.g., Pro-origami, SSDraw, iCn3D), FlatProt combines interactivity, scalability, and structural accuracy, while allowing customization and integration with SCOP/CATH classifications.
7. Benchmarking on 1,000 proteins from the human proteome shows FlatProt scales well across proteins of varying sizes (16–2,663 residues), with <6s runtime per structure on standard hardware.
8. FlatProt supports large-scale family overlay plots—e.g., for KLK proteins—enabling simultaneous visualization of 40 members in a single graphic with opacity reflecting structural conservation.
9. Designed for accessibility, FlatProt requires only Python and common bioinformatics tools (DSSP, Foldseek), and can run on typical laptops, democratizing large-scale structural comparisons.
10. By providing consistent, lightweight, and interpretable 2D protein maps, FlatProt paves the way for broader adoption of structural analysis in bioinformatics pipelines, education, and AI model training.
💻Code: github.com/t03i/FlatProt
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #BioinformaticsTools #2DVisualization #StructuralBiology #SCOP #CATH #Foldseek #SecondaryStructure #ComputationalBiology #AI4Science
1
5
27
2,743
2 May 2025
FlatProt: 2D visualization eases protein structure comparison
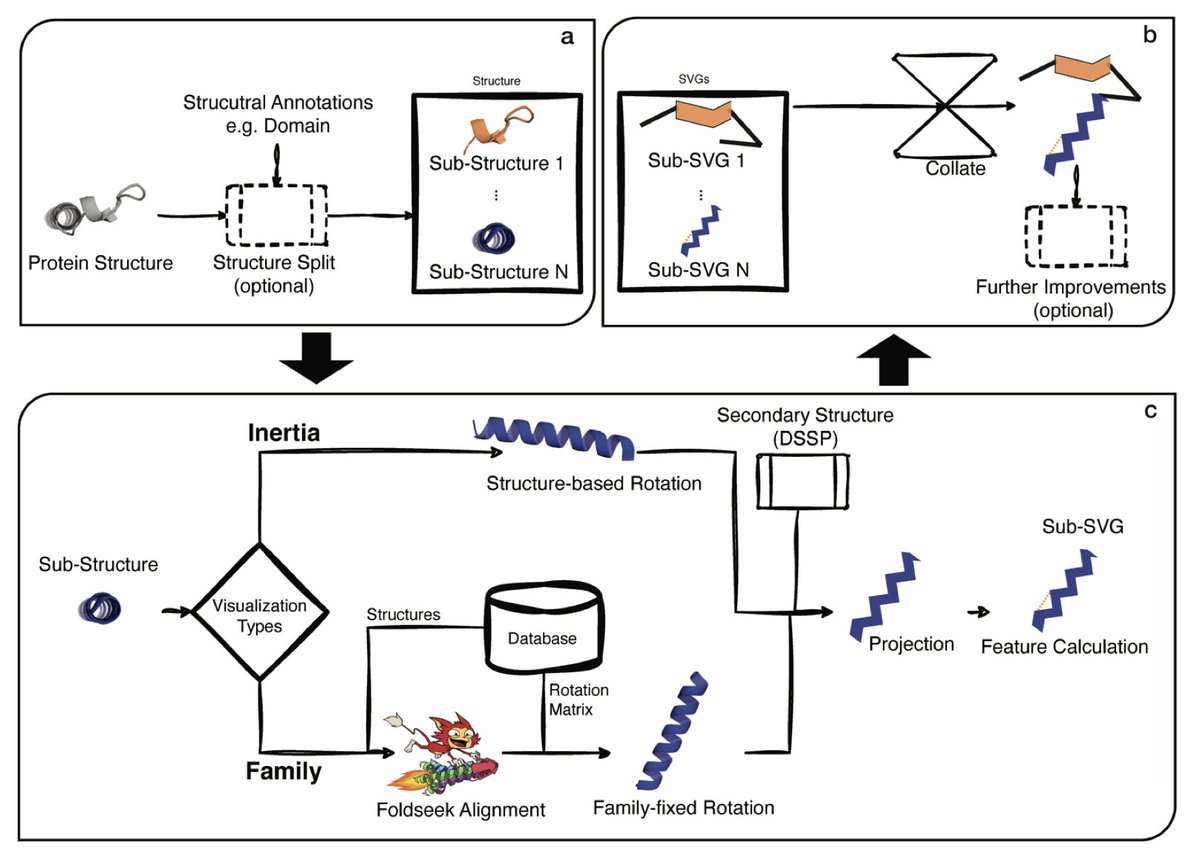
1. FlatProt is a new tool that standardizes 2D visualizations of 3D protein structures, enabling consistent, scalable, and intuitive comparison of protein families across entire proteomes.
2. The tool solves a fundamental bottleneck in structural biology: viewing and comparing many 3D protein structures on 2D screens without losing key information—an essential task in drug design and protein function analysis.
3. FlatProt employs both inertia-based and family-based structure rotation to project proteins consistently into 2D space, preserving structural orientation across related proteins and improving comparability.
4. It supports domain-aware visualizations, simplified segment representations (e.g., helices as zigzags, strands as triangles), and overlay of annotations like disulfide bonds or divergent regions via lDDT scores.
5. The tool enables consistent visualization for structurally diverse families, like Three-Finger Toxins (3FTx) and Kallikreins (KLK), making it easier to detect conserved features and family-specific variations.
6. Unlike prior tools (e.g., Pro-origami, SSDraw, iCn3D), FlatProt combines interactivity, scalability, and structural accuracy, while allowing customization and integration with SCOP/CATH classifications.
7. Benchmarking on 1,000 proteins from the human proteome shows FlatProt scales well across proteins of varying sizes (16–2,663 residues), with <6s runtime per structure on standard hardware.
8. FlatProt supports large-scale family overlay plots—e.g., for KLK proteins—enabling simultaneous visualization of 40 members in a single graphic with opacity reflecting structural conservation.
9. Designed for accessibility, FlatProt requires only Python and common bioinformatics tools (DSSP, Foldseek), and can run on typical laptops, democratizing large-scale structural comparisons.
10. By providing consistent, lightweight, and interpretable 2D protein maps, FlatProt paves the way for broader adoption of structural analysis in bioinformatics pipelines, education, and AI model training.
💻Code: github.com/t03i/FlatProt
📜Paper: biorxiv.org/content/10.1101/…
#ProteinStructure #BioinformaticsTools #2DVisualization #StructuralBiology #SCOP #CATH #Foldseek #SecondaryStructure #ComputationalBiology #AI4Science
2
6
1,916
30 Apr 2025
CS-Fold: Advancing RNA Structure Predictions through Phylogenetic Modelling of Compensatory Mutations in Deep Neural Networks
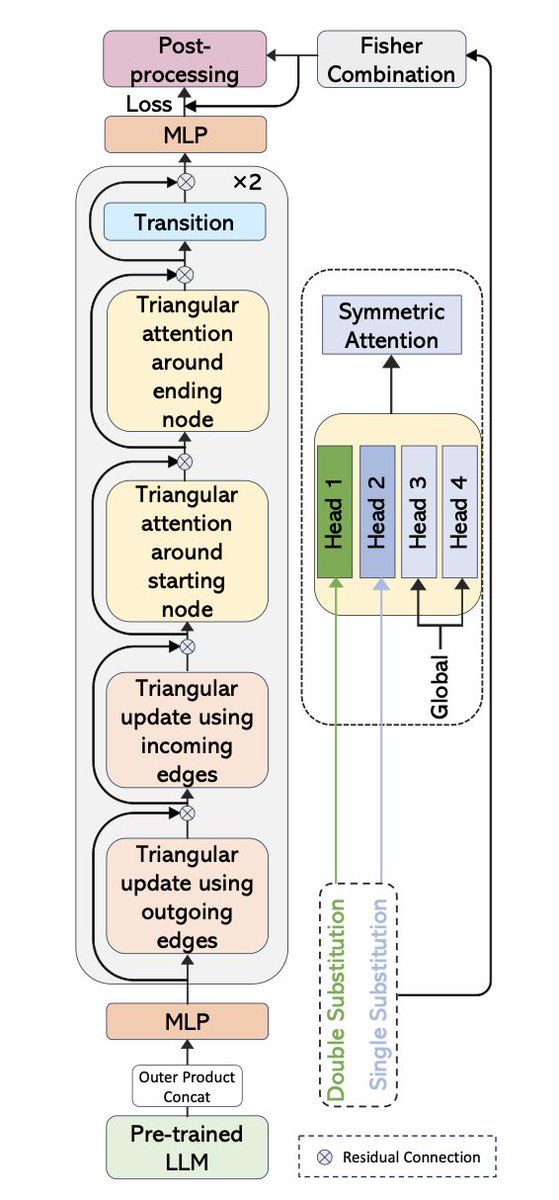
1. This study introduces CS-Fold, a deep learning framework that significantly improves RNA secondary structure prediction by explicitly modeling compensatory mutations within a phylogenetic tree—capturing co-evolutionary constraints that traditional MSA-based methods overlook.
2. The key innovation lies in integrating sparse, high-confidence compensatory substitution signals derived from reconstructed ancestral sequences across a 100-species vertebrate tree. These signals are embedded as attention biases in a Pairformer-based architecture.
3. Unlike previous approaches that treat MSAs as flat sequence alignments, CS-Fold uses likelihood-based Monte Carlo simulations over a phylogenetic tree to identify statistically significant compensatory (double) and compatible (single) substitutions.
4. The network architecture features symmetric triangular attention updates informed by compensatory mutation priors, ensuring that structural conservation across evolution guides base-pair predictions more directly and biologically meaningfully.
5. A custom loss function combines weighted binary cross-entropy, Tversky loss, and a constraint-based penalty to align predictions with substitution-derived priors, while an unrolled optimization post-processing step enforces hard constraints during inference.
6. Evaluated on a newly curated vertebrate RNA dataset comprising 13,778 sequences across 604 RNA families, CS-Fold achieves state-of-the-art performance—outperforming UFold, MXFold2, ERNIE-RNA, and Rinalmo by up to 5% in F1-score.
7. Ablation studies confirm the critical role of evolutionary priors: excluding compensatory substitution constraints from attention, loss, or post-processing significantly reduces performance, with F1-score dropping by over 18 points in the worst case.
8. CS-Fold also demonstrates strong precision, a crucial metric in RNA modeling where false positives can lead to misleading structural hypotheses—outscoring previous models while maintaining competitive recall.
9. Importantly, the model does not rely on fine-tuning large pre-trained LLMs. Instead, it uses fixed embeddings from RiNALMo and applies lightweight architectural innovations, making it efficient and generalizable even in low-data regimes.
10. By unifying deep phylogenetic modeling with neural attention mechanisms, CS-Fold offers a powerful and interpretable framework for RNA structure prediction—and sets a precedent for integrating evolutionary priors in biomolecular modeling.
📜Paper: biorxiv.org/content/10.1101/…
#RNA #SecondaryStructure #Phylogenetics #DeepLearning #ComputationalBiology #CoEvolution #AI4Science #Bioinformatics
4
13
1,007
18 Apr 2025
Predicting RNA 3D structure and conformers using a pre-trained secondary structure model and structure-aware attention
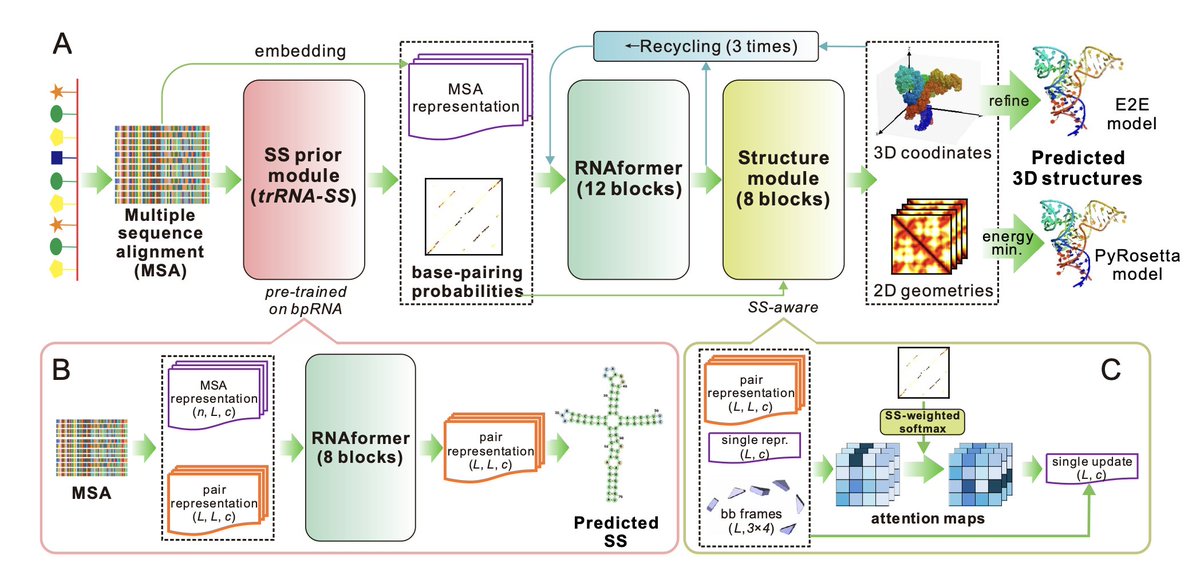
1. trRosettaRNA2 is a new deep learning model for RNA 3D structure prediction that incorporates a novel secondary structure prior module trained on bpRNA data. This allows it to overcome the data scarcity of RNA 3D structures by leveraging more abundant 2D structure information.
2. The integrated SS prior module (trRNA2-SS) not only boosts 3D prediction but also serves independently as a state-of-the-art RNA secondary structure predictor, outperforming existing tools on challenging benchmarks like ArchiveII and PDB27.
3. trRosettaRNA2 adopts an end-to-end architecture with a secondary structure-aware attention mechanism, enabling it to predict full-atom 3D structures efficiently. It achieves strong accuracy with just ~11M parameters—far fewer than models like AlphaFold3 and RoseTTAFoldNA.
4. In the CASP16 blind test, trRosettaRNA2 (as Yang-Server) ranked as the top-performing automated server group, surpassing AlphaFold3-based entries in global metrics like RMSD, TM-score, and GDT-TS, while producing structures with fewer steric clashes.
5. The model excels in modeling RNA conformational heterogeneity. On RNase P RNA, it outperformed AlphaFold3 by generating a conformational ensemble that more closely aligns with experimental AFM-derived structures (RMSF PCC = 0.894).
6. trRosettaRNA2 supports multi-source SS inputs, enabling tailored ensemble generation. This flexibility allowed it to capture diverse conformers, especially in highly dynamic regions such as the S-domain of RNase P RNA—something AlphaFold3 struggled with.
7. It is also computationally efficient. Training was completed in 12 days on a single A100 GPU, in contrast to weeks of GPU cluster usage required for competing models. Its parameter and training efficiency make it highly accessible.
8. Ablation studies reveal that the SS prior module contributes most to prediction accuracy, followed by MSA embedding. Fine-tuning strategies for reducing structural violations and improving hard targets further enhanced model quality.
9. The model’s confidence scores (pLDDT) correlate well with real RMSD, allowing for automatic model selection and quality estimation. Its recycling mechanism also compensates for inaccurate SS inputs in challenging targets.
10. trRosettaRNA2 paves the way for an "AlphaFold moment" in RNA, offering a robust, accurate, and flexible platform for RNA 3D structure and conformer prediction, while requiring significantly less computational cost than prior state-of-the-art methods.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #deeplearning #bioinformatics #RNAconformers #AlphaFold #RNAfolding #computationalbiology #AI4Science #trRosettaRNA2 #secondarystructure #structureprediction
11
47
3,001
18 Apr 2025
Predicting RNA 3D structure and conformers using a pre-trained secondary structure model and structure-aware attention
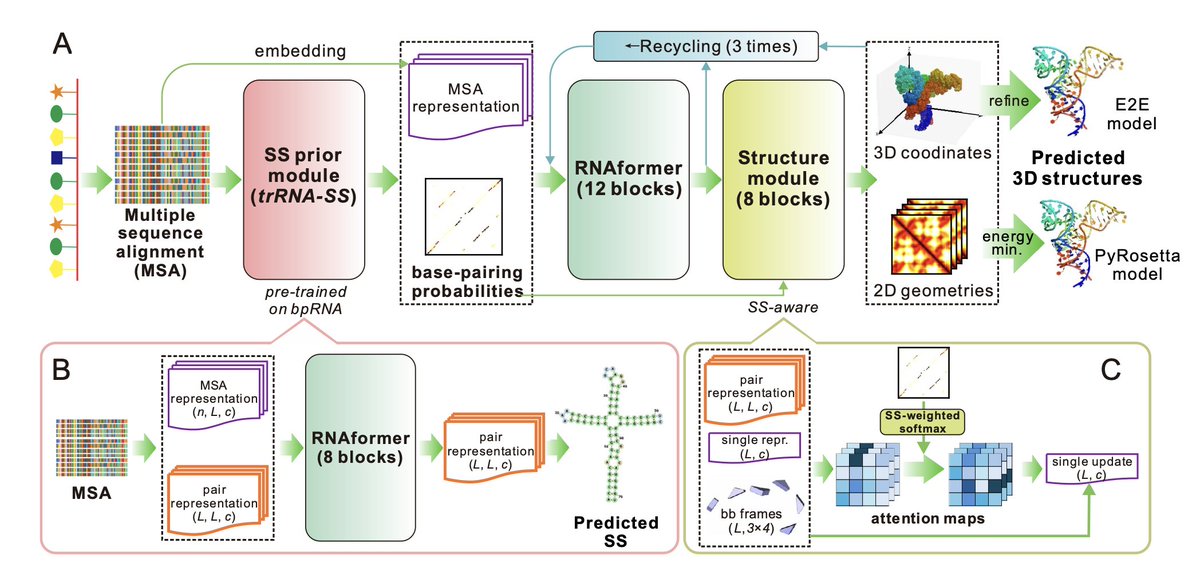
1. trRosettaRNA2 is a new deep learning model for RNA 3D structure prediction that incorporates a novel secondary structure prior module trained on bpRNA data. This allows it to overcome the data scarcity of RNA 3D structures by leveraging more abundant 2D structure information.
2. The integrated SS prior module (trRNA2-SS) not only boosts 3D prediction but also serves independently as a state-of-the-art RNA secondary structure predictor, outperforming existing tools on challenging benchmarks like ArchiveII and PDB27.
3. trRosettaRNA2 adopts an end-to-end architecture with a secondary structure-aware attention mechanism, enabling it to predict full-atom 3D structures efficiently. It achieves strong accuracy with just ~11M parameters—far fewer than models like AlphaFold3 and RoseTTAFoldNA.
4. In the CASP16 blind test, trRosettaRNA2 (as Yang-Server) ranked as the top-performing automated server group, surpassing AlphaFold3-based entries in global metrics like RMSD, TM-score, and GDT-TS, while producing structures with fewer steric clashes.
5. The model excels in modeling RNA conformational heterogeneity. On RNase P RNA, it outperformed AlphaFold3 by generating a conformational ensemble that more closely aligns with experimental AFM-derived structures (RMSF PCC = 0.894).
6. trRosettaRNA2 supports multi-source SS inputs, enabling tailored ensemble generation. This flexibility allowed it to capture diverse conformers, especially in highly dynamic regions such as the S-domain of RNase P RNA—something AlphaFold3 struggled with.
7. It is also computationally efficient. Training was completed in 12 days on a single A100 GPU, in contrast to weeks of GPU cluster usage required for competing models. Its parameter and training efficiency make it highly accessible.
8. Ablation studies reveal that the SS prior module contributes most to prediction accuracy, followed by MSA embedding. Fine-tuning strategies for reducing structural violations and improving hard targets further enhanced model quality.
9. The model’s confidence scores (pLDDT) correlate well with real RMSD, allowing for automatic model selection and quality estimation. Its recycling mechanism also compensates for inaccurate SS inputs in challenging targets.
10. trRosettaRNA2 paves the way for an "AlphaFold moment" in RNA, offering a robust, accurate, and flexible platform for RNA 3D structure and conformer prediction, while requiring significantly less computational cost than prior state-of-the-art methods.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #deeplearning #bioinformatics #RNAconformers #AlphaFold #RNAfolding #computationalbiology #AI4Science #trRosettaRNA2 #secondarystructure #structureprediction
4
15
977

26 Sep 2024
Rapid Quantification of Protein Secondary Structure Composition from a Single Unassigned 1D 13C Nuclear Magnetic Resonance Spectrum | Journal of the American Chemical Society @Yale @YaleChem #Protein #SecondaryStructure #NMR @nmr900 pubs.acs.org/doi/10.1021/jac…
3
16
3,593

7 Jun 2024
The Alpha Helix Representing the Secondary Structure of Many Proteins Is Related to the Golden Ratio Concept in at least Five Facets
~ Critique welcome ~
SG
#helix #alphahelix #doublehelix #DNA #goldenratio #protein #secondarystructure
@ResearchGate: researchgate.net/publication…
1
35

RNA Masonry automates modeling of #3Dstructures by assembling #RNA fragments based on user-provided #secondarystructure constraints, tertiary contact restraints, and small-angle X-ray scattering data. [co-authored by EVBC member @jmbujnicki] doi.org/10.1093/bioinformati…
7
490

1 Feb 2023
#HighlyCitedPaper #HighlyAccessedPaper
Title: Effect of Three Defatting Solvents on the Techno-Functional Properties of an Edible Insect (Gryllus bimaculatus) Protein Concentrate
By: Seong-Jun Cho, et al.
👉doi.org/10.3390/molecules261…
📌#insect #solventextraction #secondarystructure

2
237

12 Jan 2023
[Workshop Information]
CD measurement and secondary structure analysis of protein samples: BeStSel with Best Scene, toward Best Sailing
jasco.co.jp/jpn/event/event/…
#CircularDichroism #secondarystructure #SSE #BeStSel #protein #JASCO #日本分光
2
3
268
18 Dec 2022
Direct Correlation between the Secondary Structure of an Amphiphilic Polymer and Its Prominent Antiviral Activity @iacskolkata #SecondaryStructure #Amphiphilic #Polymer #Antiviral #Activity pubs.acs.org/doi/10.1021/jac…
5
2,955

24 Jul 2022
High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation. #DNA #RNA #SecondaryStructure #TranscriptionRegulation #TranslationRegulation @GenomeBiology #Review
genomebiology.biomedcentral.…
2
6
29 Aug 2021
Review of machine learning methods for RNA secondary structure prediction. #RNA #SecondaryStructure #MachineLearning
journals.plos.org/ploscompbi… @PLOSCompBiol
1
4

31 Jul 2021
Opinions on #AlphaFold structures like these. Is this just what proteins look like when loops are visualised or is this an artefact/unreliable #prediction? #structuralbiology #Embl #proteins #loops #folding #foldit #secondarystructure #spaghetti
1

Quick group pictures with our mini-toober Zinc Fingers @3DMolDesigns #alphahelix #betasheet #hydrogenbond #secondarystructure #weloveproteins




2
2
RNA #secondarystructure at the transcription start site influences #EBOV #transcription initiation and replication in a length- and stability-dependent manner. Paper co-authored by EVBC member S Becker @SBeckerLab
doi.org/10.1080/15476286.202…
1
2
1 Jul 2020
Fluorescence and CD Spectra using Temperature/Wavelength Measurement program
#circulardichroism #fluorescence #secondarystructure #proteins #thermodynamics #denaturation #enthalpy #entropy #JASCO
jasco-global.com/solutions/f…

3
Modeling protein #secondarystructure using #zincfinger manipulatives from @3DMolDesigns #alphahelix #betasheet




1
2