
Exploring multi-agent systems reveals how identity emerges in AI. The unique topology impacts language models beyond single interactions. #AI #Innovation #LanguageModels
2

Jun 9
The June 3, 2026 explainx.ai bulletin summarizes key AI developments, resources, and advancements in LLM technology.
👇
📖 t.me/ai_narrotor/4380
🎧 t.me/ai_narrotor/4381
#AIUpdates, #TechBulletin, #LanguageModels


The goal is not a less careful model. It's a model whose care is correctly placed - careful where care belongs, free where freedom belongs, gated by what kind of situation it is.
#AISafety #LLM #MechanisticInterpretability #AIAlignment #OverAlignment #LanguageModels
19

Jun 8
Breakthrough in Language Models: AI Just Got a LOT Smarter! New study reveals a game-changing technique to make large language models work across languages with 25% more accuracy!
arxiv.org/abs/2606.06586
#LanguageModels #AIAdvancements
2
17

May 27
✈️ Looking forward to an exciting #SIGMOD2026!
👇 Schedule below.
🇮🇳 See you soon in #Bengaluru!
#Databases #LLMs #LanguageModels #DatabaseBenchmarking #QueryOptimization #QuantumComputing
@lojil192574 @sigmod @SIGMODConf

9
409

May 18
📄 Paper: arxiv.org/abs/2602.19066
💻 Code: github.com/David-cripto/IDLM
📢 Invitation post: x.com/diffusion_llms/status/…
Many thanks to the organizers for the invitation,
@jdeschena, @ssahoo_, @zhihanyang_! 🙌
#ICML2026 #DiffusionModels #LanguageModels #GenerativeAI #MachineLearning

📢 May 18 (Mon): IDLM: Inverse-distilled Diffusion Language Models
🤔Diffusion Language Models (DLMs) have recently achieved strong results in text generation. However, their multi-step sampling leads to slow inference, limiting practical use.
💡To address this, the authors extend Inverse Distillation, a technique originally developed to accelerate continuous diffusion models, to the discrete setting. However, this extension introduces both theoretical and practical challenges.
🔧To overcome these challenges, the authors first provide a theoretical result demonstrating that their inverse formulation admits a unique solution, thereby ensuring valid optimization. They then introduce gradient-stable relaxations to support effective training.
📊As a result, experiments on multiple DLMs show that their method, Inverse-distilled Diffusion Language Models (IDLM), reduces the number of inference steps by 4×—64×, while preserving the teacher model’s entropy and generative perplexity.
This Monday, David Li (scholar.google.com/citations…) and Nikita Gushchin (scholar.google.com/citations…) will present their jointly led paper, which was recently accepted at ICML 2026.
Collaborators of this work include: Dmitry Abulkhanov (@dabulkhanov_), Eric Moulines (scholar.google.com/citations…), Ivan Oseledets (@oseledetsivan), Maxim Panov (@maxim_panov), Alexander Korotin (akorotin.netlify.app/)
Paper link: arxiv.org/abs/2602.19066

1
10
459

Building GLM-X: a graph-native cognitive architecture beyond transformers, now learning language from structured world knowledge. 👽🔥
#CognitiveAI #artificialintelligence #languagemodels #worldmodels
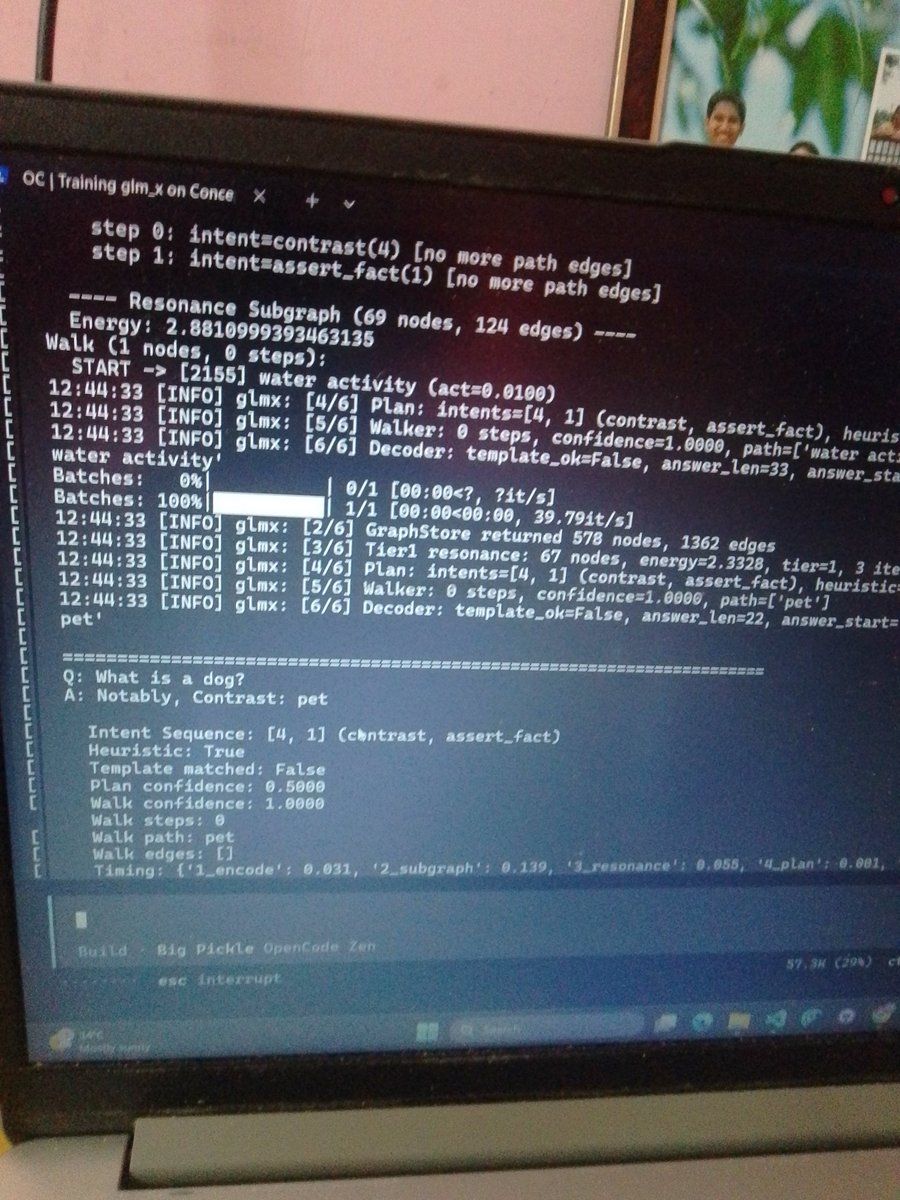
1
2
27

Our collaboration with @AnthropicAI is key to advancing safe, enterprise grade AI. Hear from @janaksevak on how Anthropic and Infosys came together in Bengaluru - training 3,000 engineers and running a hands on #hackathon with Claude models to solve real enterprise challenges at scale. Watch the video.
#EnterpriseAI #InfosysTopaz #ResponsibleAI #LanguageModels
1
5
1,492

Separating selection from mutation in antibody language models
1. The paper argues that standard masked antibody language models (e.g., AbLang2) unintentionally learn nucleotide-level biology (codon accessibility and somatic hypermutation context) and that this “mutation signal” can actively hurt prediction of functional effects of amino-acid substitutions.
2. A concrete diagnostic: when scoring substitutions at a masked site, AbLang2 assigns ~100× lower probability to amino acids that require multiple nucleotide changes from the wild-type codon versus those reachable by a single nucleotide change, revealing codon-table imprinting rather than pure functional constraint.
3. The same confounding appears at the site level: AbLang2’s amino-acid probabilities correlate strongly with neutral SHM mutability estimates across V-encoded regions, indicating the model’s outputs are entangled with mutation-rate variation independent of selection.
4. This entanglement degrades functional prediction: on a large deep mutational scan of antibody expression, AbLang2’s correlation with experimental effects drops markedly for substitutions requiring 2–3 nucleotide changes (e.g., ~0.49 for 1-nt-accessible vs ~0.30 for multi-nt-accessible), consistent with mutation bias distorting “fitness” scores.
5. The proposed solution is a factorized mutation–selection framework: a Deep Amino Acid Selection Model (DASM) learns per-site, per-amino-acid selection factors, while a separate fixed neutral nucleotide mutation model supplies codon mutation probabilities; functional effects are thus isolated in the selection term.
6. Training uses phylogeny-derived parent–child pairs from B cell clonal families (≈2 million PCPs). The likelihood of each child codon is modeled as neutral codon mutation probability over branch length t multiplied by a DASM-predicted selection factor for the corresponding amino-acid change, with joint optimization over t and DASM parameters.
7. The neutral mutation component is trained on out-of-frame (non-functional) sequences and includes a “multihit correction” to better match observed rates of 2- and 3-nucleotide codon changes, addressing SHM clustering effects that simple independent-site models underestimate.
8. Benchmarking (zero-shot) shows consistent gains: on FLAb’s largest datasets, DASM substantially outperforms AbLang2 and also beats large general protein LMs (ESM2) and an autoregressive model (ProGen2) for both binding and expression prediction, with strong correlations for heavy and light chains (e.g., expression ~0.69 heavy / ~0.67 light in one key dataset).
9. Beyond DMS, the factorized model better predicts natural affinity maturation trajectories: combining neutral mutation probabilities with DASM selection improves prediction of where nonsynonymous mutations occur and lowers “conditional perplexity” for the identities of observed mutations (median ~4.88 vs ~7.39 for AbLang2 on held-out PCPs).
10. Practical impact: the best-performing DASM is ~4M parameters (vs 45M AbLang2; 650M ESM2) and outputs selection factors for all substitutions in one forward pass, yielding major speedups (on CPU ~0.0097 s/seq vs ~11.05 for AbLang2 and ~112.6 for ESM2), enabling laptop-scale antibody mutational scanning with interpretable per-mutation selection readouts.
💻Code: github.com/matsengrp/netam ; github.com/matsengrp/dasm-ex…
📜Paper: doi.org/10.7554/eLife.109644
#ComputationalBiology #ProteinEngineering #Antibodies #MachineLearning #DeepLearning #Evolution #SomaticHypermutation #LanguageModels #Bioinformatics #Immunology

3
26
3,939
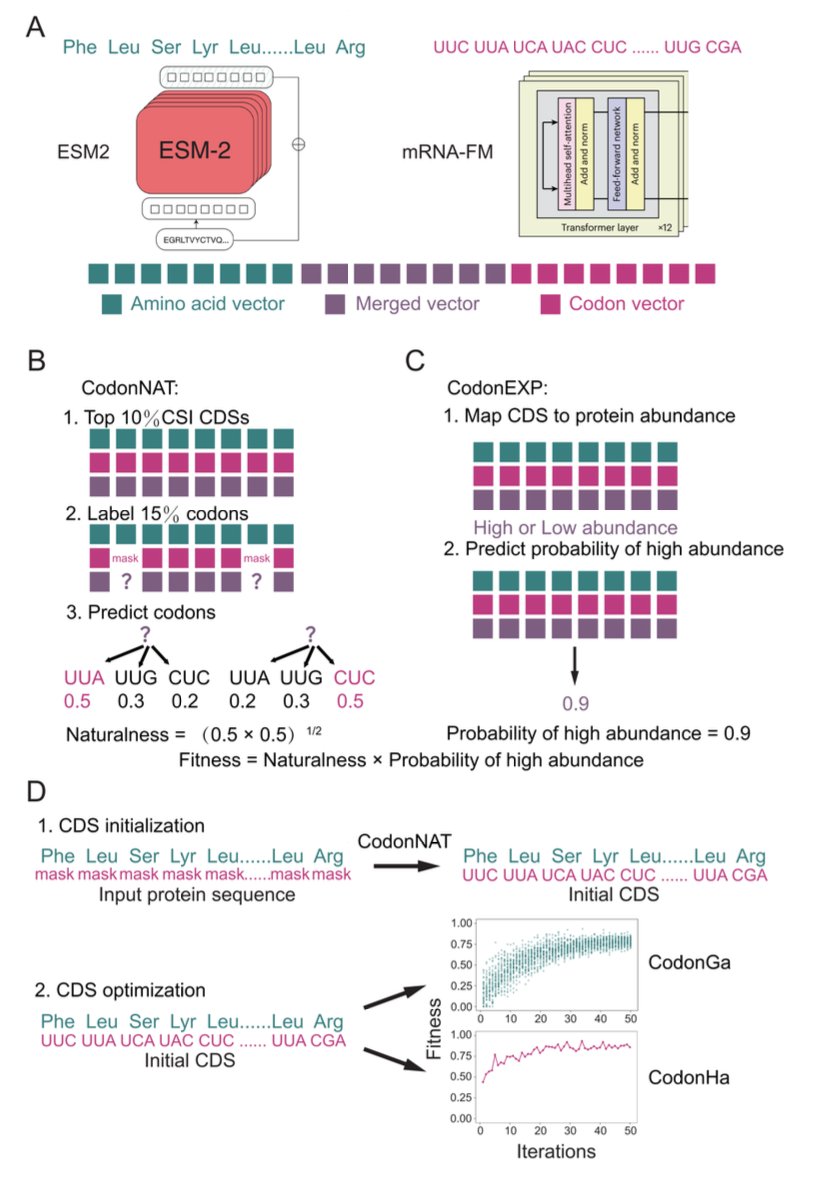
HalluCodon enables species-specific codon optimization using multimodal language models
1 HalluCodon is a plant-focused codon-optimization framework that fine-tunes pre-trained protein and RNA language models to generate species-specific coding sequences, aiming to improve heterologous protein expression beyond simple codon-frequency heuristics.
2 The core idea is a two-module scoring system: CodonNAT quantifies “codon-context naturalness” (how compatible a CDS looks relative to endogenous host genes), while CodonEXP predicts the probability that a CDS will yield high protein abundance using experimental protein abundance labels.
3 CodonNAT is built via joint fine-tuning of ESM2-650M (protein LM) and mRNA-FM (codon-token RNA LM) under a masked-language-modeling objective, learning host-specific codon context signatures rather than only per-codon frequencies.
4 Across 15 plant species (including maize, rice, tobacco, wheat, tomato, potato, grape, etc.), CodonNAT achieved higher masked-codon prediction accuracy than a “pick the most frequent codon” baseline (average 66.5% vs 56.6%), with especially strong gains for amino acids with higher synonymous-codon diversity.
5 CodonNAT also showed biologically meaningful signal in a non-plant benchmark: in E. coli ccdA synonymous-mutation fitness data, it improved correlation between predicted and measured fitness (Spearman 0.41) compared with frequency-based scoring (0.32) and slightly above CodonTransformer (0.39), supporting that it captures context effects relevant to cellular fitness.
6 CodonEXP integrates nucleotide-level and protein-level information by learning from both CDS and amino acid sequence features, supervised with protein abundance data (PaxDb-derived labels: top 33% vs bottom 33%). It reached ~79.3% average accuracy and 86.1% average AUC across the 15 plant species, and outperformed RNA-only language model baselines in maize/rice/tobacco comparisons.
7 For sequence generation, HalluCodon offers (a) a genetic algorithm (CodonGa) and (b) a hallucination-style, gradient-guided optimizer (CodonHa). Both maximize a Fitness score defined as Naturalness (CodonNAT) × Expression probability (CodonEXP), but CodonHa converges far faster in compute.
8 In a tobacco DsRed2 optimization example, CodonHa reached near-maximal predicted expression probability in only a few iterations and ran ~46.8× faster than the genetic algorithm on the reported GPU setup, while maintaining codon-context compatibility.
9 Experimental transient expression in tobacco leaves tested five proteins (DsRed2, mCry2Ab, GAT, infliximab-A, infliximab-B). For DsRed2, CodonHa produced the strongest fluorescence and higher protein levels by Western blot (reported 1.57× vs CodonTransformer, 4.32× vs Genewiz, 13.58× vs a frequency baseline), suggesting the combined NAT EXP objective can translate to wet-lab gains.
10 The study highlights GC3 as a learned and actionable plant expression feature: HalluCodon optimization tends to increase GC3 toward host-like levels, and a GC3-rewarding variant (Ha-GC3) enabled expression of larger proteins that were difficult under the default CodonHa, while warning that extreme GC3/GC increases can complicate synthesis and increase methylation-site density.
💻Code: github.com/YuxuanLou/HalluCo…
📜Paper: biorxiv.org/content/10.64898…
#CodonOptimization #PlantSyntheticBiology #ComputationalBiology #Bioinformatics #LanguageModels #DeepLearning #ProteinExpression #Transgenic #MolecularFarming #ESM2 #RNAFM
2
20
1,334

Researchers say internal emotion-like signals shape how large language models make decisions. dlvr.it/TRsjK9 #AI #ArtificialIntelligence #EmotionVectors #LanguageModels #MachineLearning
2
8

Language models have shown effectiveness in a variety of software applications, particularly in tasks related to automatic workflow.
By @languagemodels
#llms #ondevicelanguagemodels

1
2
264

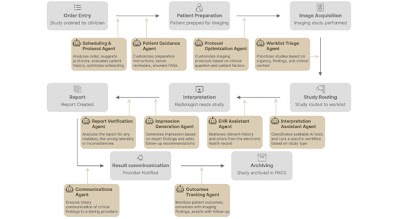
Agentic AI in Radiology: Evolution from Large Language Models to Future Clinical Integration doi.org/10.1148/ryai.250651 @judywawira @k_bressem @Tugba_Akinci_MD #agent #LLM #LanguageModels
1
4
311

Mar 31
💎 NLP Unleashed best practices revealed
New comprehensive guide covering:
✨ Core concepts
🔧 Practical examples
⚡ Performance tips
🎯 Best practices
Dive in 👇
🔗 kubaik.github.io/nlp-unleash…
#Blockchain #AR #WebDev #AIEngineering #LanguageModels
2
24
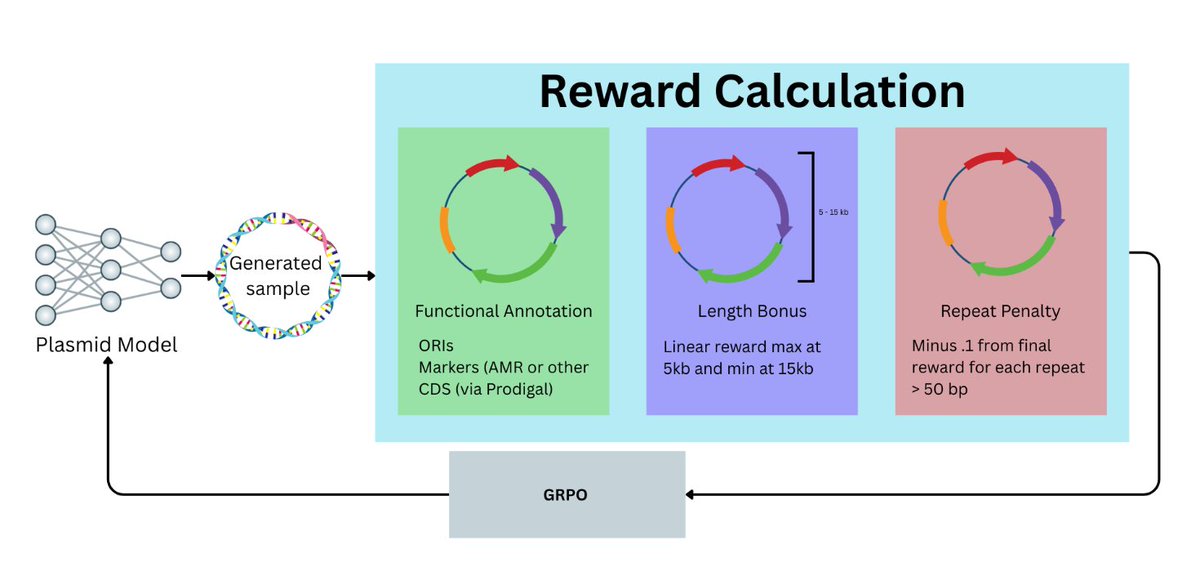
Emergent Biological Realism in RL-Trained DNA Language Models
1. The paper studies whether reinforcement learning (RL) post-training can steer a DNA language model toward “biological realism” in plasmid generation, using plasmids as a tractable testbed with clear functional constraints and major biotech relevance.
2. Using Group Relative Policy Optimization (GRPO) on top of PlasmidGPT with a biologically motivated reward, the RL model reaches a 77% in silico QC pass rate vs 5% for the pretrained baseline (and 10% for supervised fine-tuning), across both weak prompts (single “ATG”) and structured prompts (e.g., a GFP cassette).
3. The reward is intentionally lightweight and interpretable: (i) functional annotation scoring (ORI, selectable markers, promoters/terminators, CDS) with an added promoter→CDS→terminator cassette organization bonus, (ii) a length prior favoring typical 5–15 kb plasmids, and (iii) a penalty for long exact repeats (≥50 bp) linked to instability/recombination.
4. A key observation: beyond what is explicitly rewarded, the RL model’s samples align with real engineered plasmids on multiple distributional properties that were not directly optimized—thermodynamic stability (MFE density), codon usage patterns, and ORF length distributions—suggesting correlated “realism” emerges when optimizing for a subset of structural constraints.
5. Quantitatively, RL generations are closest to real plasmids in GC content (mean 0.518 vs real 0.517; matched variance), 3-mer/codon composition similarity (lowest mean Jensen–Shannon divergence to real), and length/stability distributions (median length 6668 vs real 6690; reduced variance; MFE density mean close to real with much smaller variance than base/SFT).
6. Novelty is reduced but remains substantial: 67% of RL generations are classified as novel by BLASTn thresholds (vs 91% base, 96% SFT). Importantly, RL yields many more sequences that are both QC-passing and novel (60% of all RL samples), indicating improvement is not explained purely by memorizing known plasmids.
7. RL concentrates probability mass (diversity drops from 0.915 to 0.588 by pairwise 21-mer Jaccard distance), consistent with learning conserved “successful motifs” (e.g., reliable ORIs/markers) without collapsing to identical outputs; the paper frames this as a practical quality–diversity trade-off.
8. Unlike the common “alignment tax” seen in NLP RLHF, RL post-training does not degrade next-token prediction on held-out continuation; it slightly improves mean log-probability (base −12.449 vs RL −10.966) and substantially reduces variance, suggesting more consistent modeling while being optimized at the sequence level.
9. The paper also probes reward hacking concerns: CDS-related reward uses Prodigal (statistical gene prediction, not homology). The RL model shows lower CDS surprisal on real plasmids than the pretrained/SFT models, supporting the interpretation that RL sharpened general plasmid-like structure rather than overfitting to quirks of the reward.
10. Limitations and outlook: evaluation is bioinformatics-only (library-dependent, potentially penalizing truly novel functional parts like unseen ORIs), and wet-lab validation is not performed here. The authors argue this is groundwork for conditional plasmid design via richer prompts (host, copy number, expression goals) to recover diversity and improve practical utility.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Genomics #SyntheticBiology #DNA #LanguageModels #ReinforcementLearning #RLHF #Bioinformatics #PlasmidDesign #MachineLearning
5
11
1,555
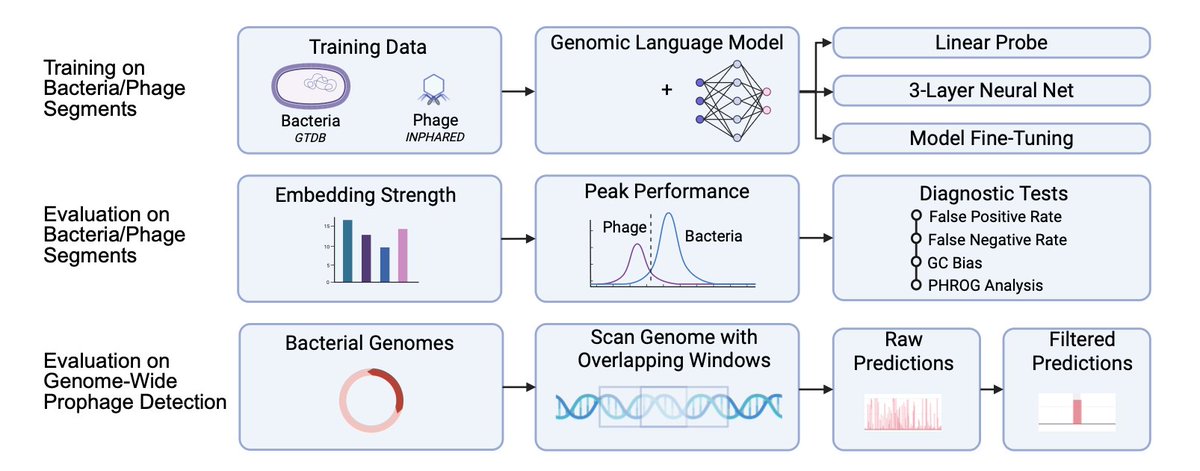
LAMBDA: A Prophage Detection Benchmark for Genomic Language Models
1 LAMBDA introduces a bacterial-domain benchmark that asks whether genomic language models (gLMs) learn whole-genome sequence features, using phage vs bacteria discrimination and genome-wide prophage localization as the central stress test.
2 The benchmark is organized into four tiers of increasing realism: (i) probing tasks on frozen embeddings, (ii) fine-tuning for peak performance, (iii) diagnostic tests (GC bias, FPR/FNR asymmetry, PHROG functional strata), and (iv) genome-wide prophage detection by scanning complete bacterial genomes with overlapping windows.
3 A key design choice is measuring representational value directly: LAMBDA compares probes trained on pretrained embeddings vs the same architectures with random weights, reporting ΔMCC. Across models, pretraining yields large gains, and non-linear probes add only modest improvements over linear probes—suggesting strong models make phage/bacteria largely linearly separable in embedding space.
4 LAMBDA evaluates diverse gLMs spanning architectures, scale, tokenization, and training corpora (e.g., ProkBERT, DNABERT-2, Nucleotide Transformer v2, GENERanno, megaDNA, Caduceus, EVO2), enabling controlled conclusions about what matters for bacterial annotation tasks.
5 Results emphasize training data relevance over sheer parameter count: models trained on prokaryotic/phage-heavy corpora (e.g., ProkBERT-mini, GENERanno) perform strongly despite being far smaller than frontier-scale models, while models trained predominantly on human DNA (e.g., DNABERT-2, Caduceus) lag on bacterial prophage tasks.
6 Diagnostic tests show GC-composition explains only a small fraction of performance (near-zero MCC on GC-preserving shuffled sequences), but error modes differ sharply across models: fragment-level false positive rates can be extremely high for some architectures (e.g., strong “phage” bias), motivating per-model and per-task diagnostics rather than relying on a single aggregate score.
7 Functional validation uses PHROG categories on phage CDS sequences, showing sensitivity varies by gene class: “Head & Packaging” and “Tail” genes are easiest across models, while “Integration & Excision” and “No PHROG match” are harder—highlighting where sequence representations capture canonical phage biology vs where “viral dark matter” remains challenging.
8 Genome-wide prophage detection is substantially harder than balanced fragment classification: raw window predictions inflate false positives, especially due to “hard negatives” like genomic islands/ICEs and degraded prophage remnants. LAMBDA therefore includes a signal extraction pipeline (per-genome z-score normalization, bidirectional EMA smoothing, clustering/merging, length and score filtering) that improves MCC with minimal recall loss.
9 On 80 bacterial genomes with 386 verified prophage regions, the best gLM (EVO2) reaches region-level MCC ~0.680 after filtering, while curated smaller models remain competitive (e.g., ProkBERT-mini and Nucleotide Transformer v2 ~0.658; GENERanno ~0.648). Traditional tools still lead overall (e.g., geNomad MCC ~0.794; PHASTER ~0.786), clarifying the current gap between gLMs and homology/hybrid pipelines.
10 LAMBDA also evaluates interpretability claims: EVO2’s sparse autoencoder “prophage feature” (f/19746) fires cleanly in some genomes but not others; after clustering it is competitive yet below an EVO2 classifier (MCC ~0.636 vs ~0.680), suggesting prophage signal may be domain-dependent and/or distributed across circuits rather than captured by a single feature.
💻Code: github.com/leannmlindsey/LAM…
📜Paper: biorxiv.org/content/10.64898…
#Genomics #Bioinformatics #ComputationalBiology #MachineLearning #DeepLearning #LanguageModels #Bacteriophage #Prophage #Microbiome #Benchmarking
3
10
1,226

Mar 18
Small Language Models are proving that efficiency can also enhance cybersecurity.
A recent study, “Small Language Models for Phishing Website Detection” by Georg Goldenits and colleagues, explores how compact language models can be used to identify phishing websites, one of the most persistent threats in today’s digital ecosystem.
Instead of relying on heavy machine-learning pipelines that demand extensive feature engineering and constant retraining, the research demonstrates that lighter language models can analyze textual patterns in URLs and web content to detect phishing attempts effectively.
What makes this particularly interesting is the broader implication: smaller models are becoming powerful tools not just for language understanding but also for practical, real-world applications like cybersecurity.
When structured well, small models can deliver big impact.
Read more here > - mdpi.com/2624-800X/6/2/48
@GeorgGoldenits @AbujaHausa @bbchausa @abujahashtags @IgboLand_ @IgboHistoFacts
@EqualyzAI #SmallLanguageModels #CybersecurityAI #EfficientAI #PhishingDetection #AIResearch #LanguageModels

2
34
131

Mar 13
Researchers, including Benjamin Bogenberger, developed a robot that combines #LanguageModels with #3Dvision to locate misplaced objects by building a spatial map and estimating likely locations: go.tum.de/730486
#Robotics #AI
📷A. Schmitz
2
12
1,175
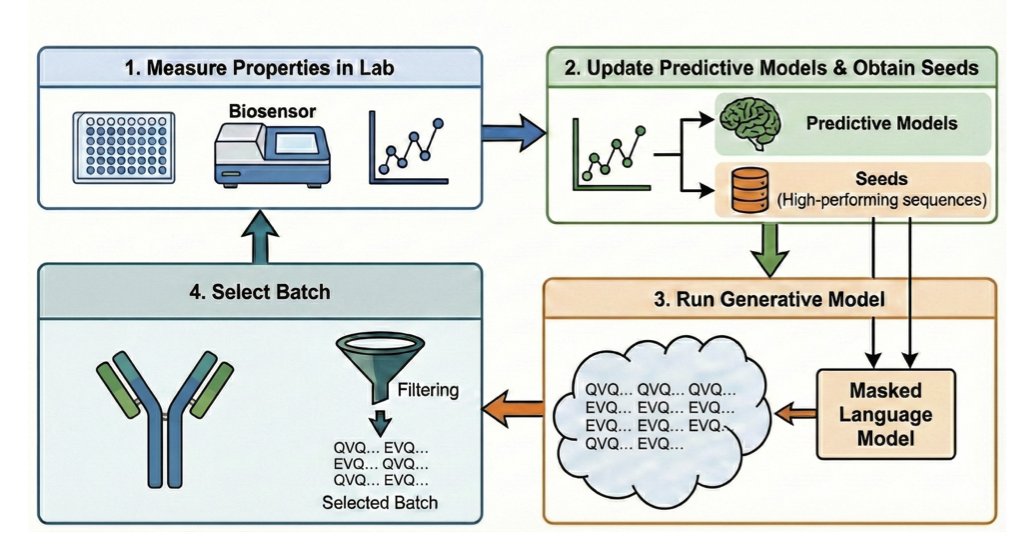
How to Make the Most of Your Masked Language Model for Protein Engineering
1 The paper introduces a new sampling method for masked language models (MLMs) in protein engineering that significantly outperforms existing approaches, with the key finding that choice of sampling algorithm matters at least as much as choice of model.
2 The core innovation is temperature-annealed stochastic beam search, which reframes generation as sequence evaluation rather than mutation-centric sampling, exploiting MLMs' efficiency at evaluating pseudoperplexity of entire 1-edit neighborhoods.
3 This sequence-centric approach enables flexible multi-objective optimization with black-box scoring functions, overcoming limitations of mutation-centric methods that struggle with non-differentiable scores like OASis percentile or isoelectric point.
4 The method achieves dramatic computational speedups: O(BEL³) complexity compared to O(EL³) per sequence for traditional approaches, yielding 20EL× speedup for realistic scenarios while producing higher-quality sequences.
5 Extensive in vitro experiments on actual antibody therapeutics campaigns reveal that ESM-2 (trained on generic proteins) and AbLang-2 perform exceptionally well, with beam search consistently outperforming Gibbs sampling across all models.
6 When combined with supervised guidance using smooth Tchebycheff scalarization, the approach achieved 100% success rate in synthesizability and binding QC criteria, while Pareto-based non-dominated sorting showed competitive but slightly lower performance.
7 The work provides practical recommendations: use supervision when possible, prefer AbLang-2 or ESM-2-650M for antibody engineering, adopt stochastic beam search over Gibbs sampling, and carefully consider scalarization methods for multi-objective guidance to avoid undesirable side effects.
📜Paper: arxiv.org/abs/2603.10302
#proteinengineering #antibodydesign #machinelearning #bioinformatics #computationalbiology #drugdiscovery #languagemodels #MLM #beamsearch #multiobjectiveoptimization
7
33
2,972

Feb 25
“The reviewer misunderstood my paper.”
This happens more often than we like to admit—and it’s tricky to respond well.
Here’s how to handle it *without sounding defensive* 👇
---
### First: pause before pushing back
If a reviewer misunderstood something, ask yourself:
👉 *Was it truly obvious in the paper?*
---
### How to respond in the rebuttal
**Acknowledge first.**
Start with:
> “We thank the reviewer for pointing this out.”
This immediately lowers friction.
---
**Then clarify—with pointers.**
* Restate the key idea in simpler terms
* Point to exact sections, figures, or equations
* If needed, add a short explanation directly in the rebuttal
Example:
> “We have clarified this in Section 3.2 and added an illustrative example in Appendix B.”
---
### When *not* to just point back
Avoid saying:
> “This is already explained in the paper.”
Instead:
* Re-explain briefly
* Then say where it can be found
* And improve the paper wording if possible
Repetition is better than resistance.
---
### Strengthen the paper to prevent repeat confusion
If one reviewer misunderstood, others might too.
Good fixes include:
* Moving key assumptions earlier
* Adding a schematic or toy example
* Explicitly stating what your method *does not* do
---
A misunderstanding isn’t a failure—it’s feedback on communication.
Respond with humility, clarity, and evidence, and reviewers are far more likely to reconsider their stance.
___
Have a paper you've been trying to publish in #GenerativeAI #Multimodal #LanguageModels #Agents ? Check out lnkd.in/gRTE4vEv (GRAIL-V) at @CVPR. Papers due soon 🗓March 5,2026.
#Publishing #PaperWriting #ResearchTips #AcademicPublishing #Conference #Research #ConversationalAI #AgentSystems #Evaluation #Accuracy #Memory #MultiTurn #ShortTerm #LongTerm #Reliability #ACL #EMNLP #NeurIPS #ICML #ICLR #GrailV #CVPR2026

3
76