
DeepRHP: A Hybrid Variational Autoencoder for Designing Random Heteropolymers as Protein Mimics
1. The paper introduces DeepRHP, a semi-supervised hybrid VAE that learns latent representations of random heteropolymer (RHP) sequence ensembles while explicitly constraining the latent space to reflect function-related chemical features, aiming to make RHP design more data-driven than empirical screening.
2. Key architectural idea: a classical sequence VAE is paired with a parallel feature-based VAE that reconstructs a deterministic chemical feature y derived from the same sequence x; both branches share the same latent variable z, encouraging z to encode both sequence-pattern statistics and chemically meaningful structure.
3. The training objective modifies the standard VAE ELBO by combining two reconstruction terms: (a) discrete sequence reconstruction (cross-entropy over monomer tokens) and (b) feature reconstruction (MSE on y), weighted by a tunable α, while keeping the KL regularization on q(z|x) vs p(z).
4. The “feature” used for semi-supervision is the sliding-window average hydrophilic–lipophilic balance (HLB), motivated by prior evidence that local hydrophobicity/solubility patterning is strongly tied to RHP behavior in protein stabilization and transport applications.
5. Data pipeline: the study simulates 10,000 RHP sequences per monomer composition using Compositional Drift (copolymer models Monte Carlo), focusing on a 4-methacrylate monomer set (MMA, EHMA, OEGMA, SPMA) spanning hydrophobic, very hydrophobic, hydrophilic, and charged chemistries.
6. To connect synthetic polymers to biology, ~30k membrane and ~30k globular protein sequences (UniProt, 50% identity threshold) are reduced into a 4-letter “monomer-equivalent” alphabet based on residue hydrophobicity/charge, enabling joint embedding and similarity analysis between proteins and RHP ensembles.
7. Design insight 1 (alphabet size): by comparing 2-monomer vs 4-monomer RHP libraries in the learned latent space (visualized via PCA of latent factors), the paper argues that 2-monomer sequence space is too broad relative to protein-like regions, whereas 4-monomer libraries yield more localized, protein-overlapping distributions—supporting why four monomers can be “enough” for protein-mimic behavior.
8. Design insight 2 (composition): within a fixed 70% hydrophobic / 30% hydrophilic constraint, varying the MMA:EHMA ratio produces distinct RHP ensembles; DeepRHP’s latent-space overlap with Aquaporin Z (AqpZ) projections highlights specific compositions (notably matching the published optimal formulation) as most similar to the target membrane protein.
9. Practical takeaway: DeepRHP reframes RHP design as an ensemble-level representation learning problem—enabling composition suggestion by latent-space similarity to target proteins—without requiring exact polymer sequences, 3D structures, or multiple sequence alignment, and with a plug-in pathway to incorporate other chemical features beyond HLB.
10. The authors report ablations indicating the hybrid (feature-guided) architecture outperforms a classical VAE alone for producing useful latent structure, while noting that current evaluation is largely qualitative and motivating future quantitative metrics and downstream tasks (e.g., membrane protein subclass discrimination, RHP–protein similarity scoring).
📜Paper: arxiv.org/abs/2606.11651
#ComputationalBiology #MachineLearning #DeepLearning #VAE #GenerativeModels #PolymerScience #MaterialsInformatics #ProteinEngineering #MembraneProteins #Cheminformatics

2
14
1,118

Jun 11
Riken Technos: The Invisible Architecture youtu.be/YVKVMCtnwYQ?si=lg8z… 来自 @YouTube #RikenTechnos #PricingPower #FY2026 #EquityResearch #MaterialsInformatics #B2BStrategy #ChemicalIndustry #StockAnalysis
2
Jun 11
RIKEN TECHNOS: How to Print Money with 87.7% Profit Growth in a Stagnant... youtu.be/OfwT_MDh1ro?si=-Kxr… 来自 @YouTube #RikenTechnos #PricingPower #FY2026 #EquityResearch #MaterialsInformatics #B2BStrategy #ChemicalIndustry #StockAnalysis
3

Jun 9
FTIR deep learning → biofuel composition analysis with zero sample destruction.
Published in Results in Engineering — shorter feedback loops, higher throughput for R&D teams developing next-gen energy materials.
mi-6.co.jp/news/20260324/
#MaterialsInformatics #Biofuel
2
4
496

Jun 4
📝 Happy to write a News & Views article (doi.org/10.1038/s43588-026-0…) for the paper on the Open Materials 2024 (OMat24) inorganic materials dataset and models from the @AIatMeta FAIR Chemistry Team (@OpenCatalyst) just published in @NatComputSci.
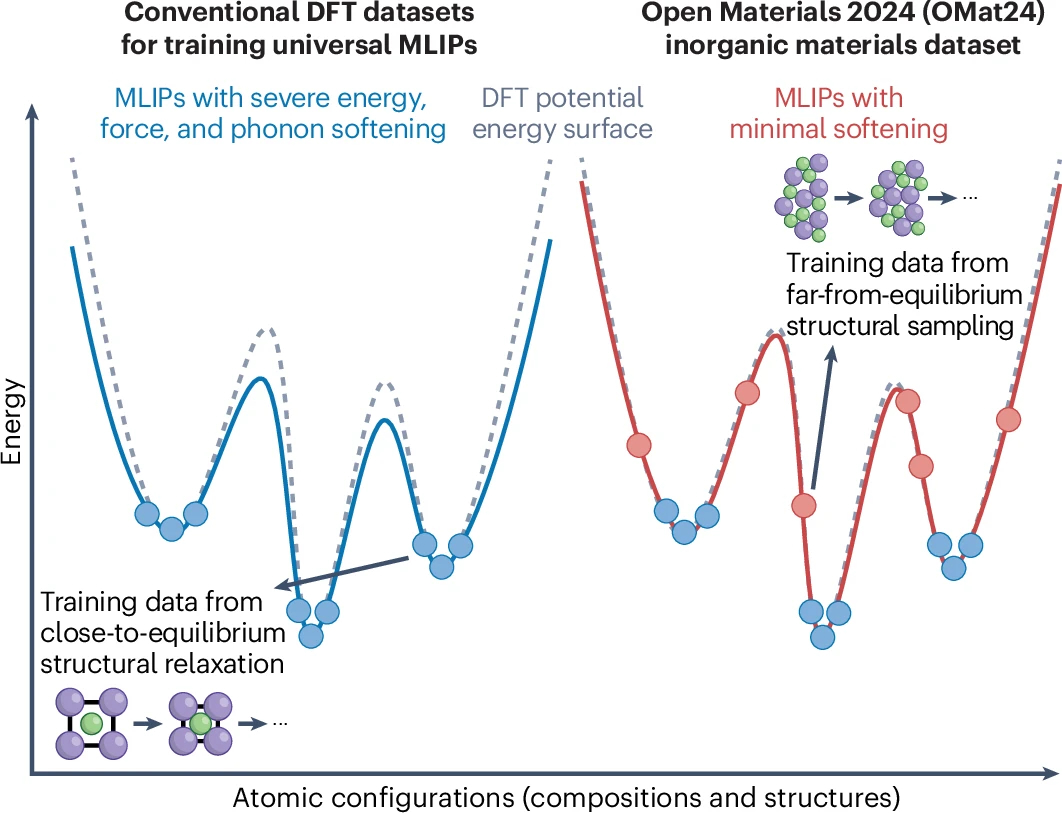
Many data-driven atomistic simulations require AI models to describe materials beyond ideal, near-equilibrium structures. At high temperature, near defects, or under realistic processing conditions, atomic environments can become strongly distorted and far from equilibrium.
This is why OMat24 (doi.org/10.1038/s43588-026-0…) is a very helpful contribution: it is one of the largest-scale materials-centric open efforts aimed at making universal machine learning interatomic potentials (MLIPs) more reliable precisely when materials are driven out of their equilibrium “comfort zone.”
This connects closely to the systematic softening problem highlighted by @Bowen_D_ and co-workers in their earlier 2025 work in npj Computational Materials (doi.org/10.1038/s41524-024-0…), where MLIPs can underestimate energies, forces, and phonons for distorted structures. OMat24 addresses this issue in a data-centric way, by expanding training data to include large-scale off-equilibrium atomic configurations so that models can better learn how materials respond when they are strongly perturbed.
The impact of OMat24 is already visible from how quickly it has become a central and widely used resource for the universal MLIP community, including benchmarking efforts on the Matbench Discovery leaderboard (matbench-discovery.materials…). To me, this work is a timely reminder that progress in atomistic machine learning depends not only on model architectures, but also on the physical coverage and design of the datasets used to train them.
Looking ahead, I am excited to see how these ideas extend toward more realistic materials chemistry, including defects, surfaces, reactive interfaces, magnetic and charge-dependent systems, higher-fidelity DFT data and methods, and more data-efficient ways to train and reuse large MLIP models and materials informatics datasets.
Many thanks to @JPanPanJ for the invitation. Congratulations to @zackulissi, @mshuaibii, @xiangfu_ml, and others in the FAIR Chemistry Team on this important contribution to the AI for Materials research community. 🎉
#MaterialsInformatics #MachineLearning #AIforScience #OpenScience

An accompanying News & Views for this paper by @peng_jiayu is also available! nature.com/articles/s43588-0…
🔓rdcu.be/fl8fo
16
1,722

May 4
📚 𝗥𝗲𝘀𝗲𝗮𝗿𝗰𝗵 𝗣𝗮𝗽𝗲𝗿𝘀 𝘄𝗲 𝗹𝗼𝘃𝗲
Two papers we found especially interesting this week:
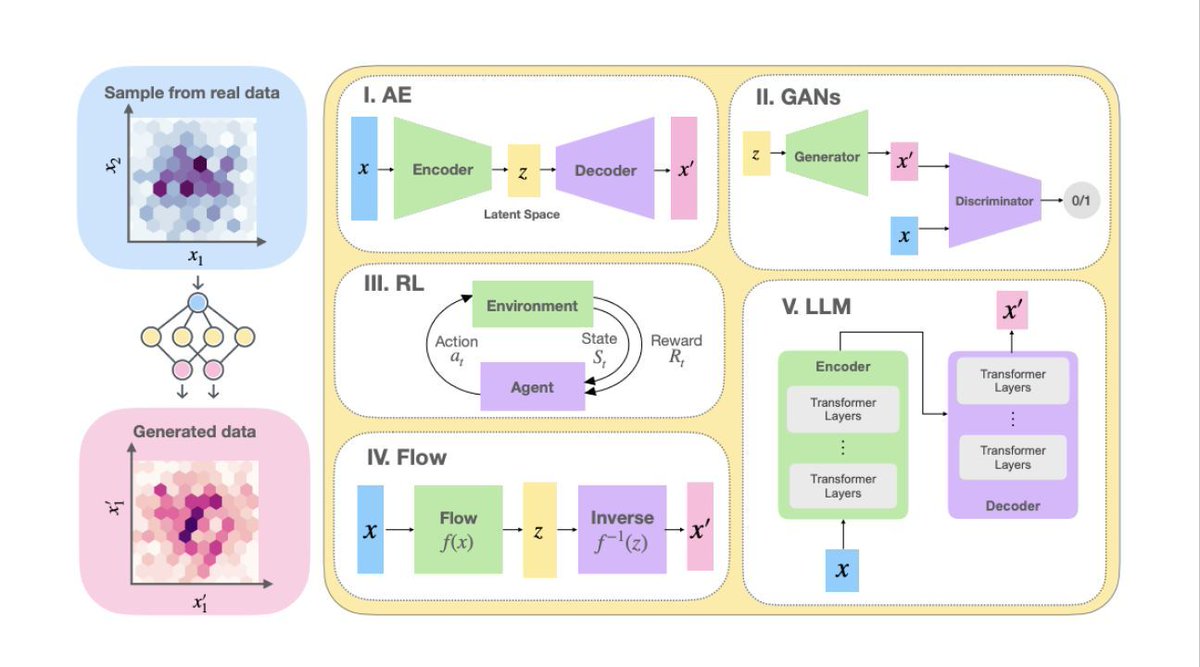
🧪 𝗚𝗲𝗻𝗲𝗿𝗮𝘁𝗶𝘃𝗲 𝗮𝗿𝘁𝗶𝗳𝗶𝗰𝗶𝗮𝗹 𝗶𝗻𝘁𝗲𝗹𝗹𝗶𝗴𝗲𝗻𝗰𝗲 𝗳𝗼𝗿 𝗰𝗼𝗺𝗽𝘂𝘁𝗮𝘁𝗶𝗼𝗻𝗮𝗹 𝗰𝗵𝗲𝗺𝗶𝘀𝘁𝗿𝘆
This paper looks at how generative AI is advancing molecular sampling, force field development, and simulation speed in computational chemistry. Its key point is important: scientific AI should not only fit known patterns, but also help predict emergent chemical phenomena. The authors also argue that future models may need deeper integration with statistical mechanics.
🔗 Source: arxiv.org/pdf/2409.03118
🔬 𝗙𝗼𝘂𝗻𝗱𝗮𝘁𝗶𝗼𝗻𝗮𝗹 𝗟𝗮𝗿𝗴𝗲 𝗟𝗮𝗻𝗴𝘂𝗮𝗴𝗲 𝗠𝗼𝗱𝗲𝗹𝘀 𝗳𝗼𝗿 𝗠𝗮𝘁𝗲𝗿𝗶𝗮𝗹𝘀 𝗥𝗲𝘀𝗲𝗮𝗿𝗰𝗵
This paper introduces LLaMat, a family of domain-adapted foundation models for materials science. The results show strong performance in materials-specific NLP, structured information extraction, and crystal structure generation. One of the most useful takeaways: domain adaptation still matters. Bigger general-purpose models are not automatically better for scientific work.
🔗 Source: arxiv.org/abs/2412.09560
Taken together, these papers point to a broader shift: scientific AI is becoming more predictive, domain-aware, and practically useful for chemistry and materials research.
#ResearchPapers #AIforScience #MaterialsScience #ComputationalChemistry #ComputationalMaterials #GenerativeAI #LLM #MaterialsInformatics
5
255

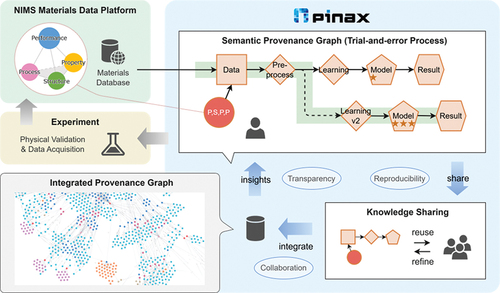
#STAMMethods "pinax" streamlines data workflows and provenance for materials science, enabling reproducibility and efficient knowledge sharing.
🔗 doi.org/10.1080/27660400.202… #MaterialsInformatics #EditorsChoice
1
2
111
Tabular foundation models for in-context prediction of molecular properties
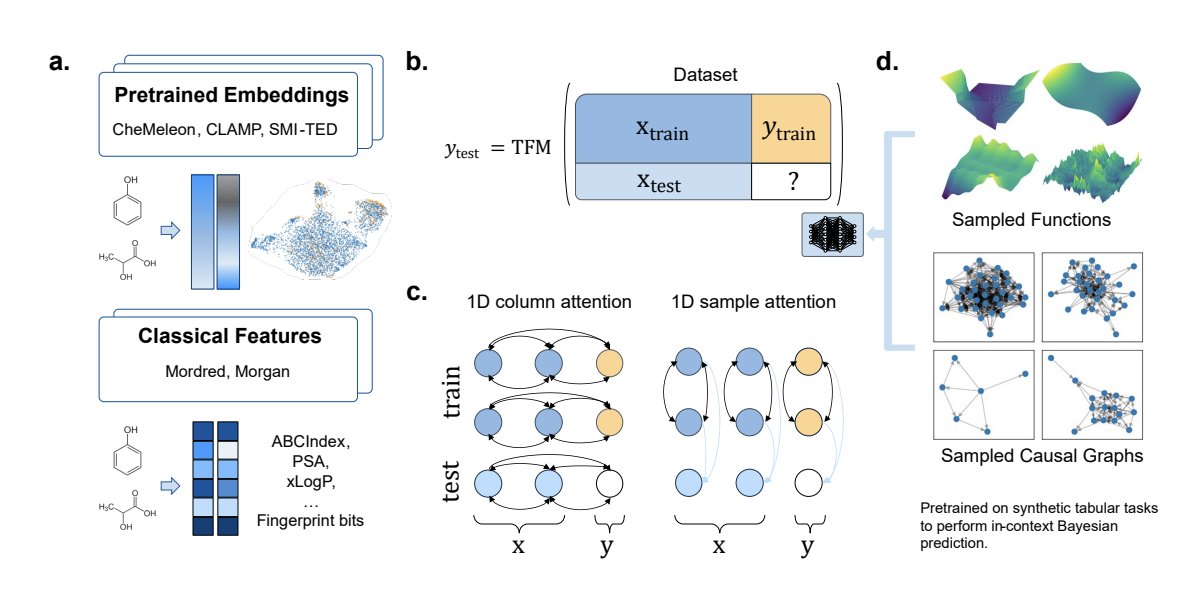
1. The paper evaluates a training-free workflow for molecular property prediction: compute fixed molecular features/embeddings, then use tabular foundation models (TFMs) for in-context learning (no task-specific fine-tuning).
2. Key result on public low-to-medium data benchmarks (58 tasks total from Polaris MoleculeACE): TabPFN frozen CheMeleon embeddings (TabPFN-CheMeleonFP) achieves 50/58 wins (86.2% win rate) with average rank 4.52, outperforming classical ML baselines and several fine-tuned deep molecular models under matched splits/metrics.
3. On the activity-cliff focused MoleculeACE suite (30 tasks), TabPFN-CheMeleonFP is best or statistically tied for best on all 30 tasks (100% win rate; average rank 2.10). This suggests TFMs can be particularly strong in difficult generalization regimes when paired with the right representation.
4. The study’s framing is notable: TFMs (TabPFN, TabICL) are pretrained only on synthetic tabular tasks (SCM/function-sampled), yet transfer effectively to chemistry once molecules are converted into tabular vectors (descriptors or frozen foundation-model embeddings).
5. Representation choice is a major driver of performance (contrary to some prior claims of representation invariance for TabPFN in drug discovery). CheMeleon embeddings and 2D descriptors (RDKit2d, Mordred) are consistently strong; Morgan fingerprints are substantially weaker across many tasks.
6. Descriptor-based alternatives remain competitive: TabPFN-RDKit2d and TabPFN-Mordred deliver strong aggregate results (e.g., 56.9% and 67.2% win rates respectively across the 58 tasks), offering practical options when foundation-model embeddings are unavailable or costly.
7. Compute efficiency is a central advantage: in a runtime case study vs fine-tuned CheMeleon, TabPFN-CheMeleonFP is faster on both CPU and GPU, with speedups up to 27× (CPU) and 46× (GPU), while also improving accuracy.
8. Beyond pharma benchmarks, the paper tests 11 chemical engineering datasets (fuels, polymer properties, polymer–solvent interactions). TFM pipelines (especially with Mordred or RDKit2d) match or exceed strong literature baselines on multiple targets, and remain competitive even when specialized models lead (e.g., PolySolv).
9. Limitations and open directions: scaling to larger datasets can become memory/disk constrained for high-dimensional features; most experiments are single-molecule (mixtures/multi-component systems may be harder); TFMs are single-task by design, motivating future in-context multitask TFMs and uncertainty-aware workflows (active learning/Bayesian optimization).
💻Code: git.rwth-aachen.de/avt-svt/p…
📜Paper: arxiv.org/abs/2604.16123
#ComputationalChemistry #Cheminformatics #MolecularML #FoundationModels #InContextLearning #TabPFN #TabularData #DrugDiscovery #ChemicalEngineering #MaterialsInformatics
1
9
1,464

Apr 21
University of Bayreuth | PhD in Materials Informatics & AI for Sustainable Polymers 🧪🤖
🚨 Deadline: Apply ASAP (rolling / until filled)
Join the University of Bayreuth and work on AI-driven design of sustainable polymer materials for electronics and packaging.
📌 Position Details
🎓 Role: Graduate Researcher / PhD Candidate (m/f/d)
🏫 Department: Kuenneth Group (Computational Materials Sciences)
👨🏫 Supervisor: Prof. Christopher Künneth
📍 Location: Bayreuth, Germany
⏳ Duration: Up to 3.5 years
💰 Salary: TV-L E13 (100% funded)
🔬 Research Focus
This position is part of an EU Horizon Europe project, combining AI, materials science, and sustainability to design next-generation polymer materials.
You will:
• 🤖 Develop machine learning models, LLMs & AI agents for materials research
• 📊 Extract, structure & manage datasets for ML training
• 🔗 Bridge AI models with experimental workflows
• 🧪 Work on sustainable polymers for electronics & packaging
• 🚀 Conduct independent research in a fast-paced environment
🌍 Project Highlights
• Work on AI materials science sustainability
• Part of a prestigious EU-funded research project
• Strong collaboration with industry & international partners
• Opportunity to contribute to real-world sustainable solutions
🎯 Ideal Candidate
• Master’s in Engineering / Computer Science / Natural Sciences
• Strong Python programming skills 💻
• Interest in materials science AI/ML
• Curious, independent, and proactive mindset
🌟 Why Apply?
• Fully funded PhD with competitive German salary (TV-L E13) 💰
• Opportunity to work with cutting-edge AI technologies (LLMs, agents)
• Flexible working hours remote work options
• Access to strong academic & industrial network
• Supportive and collaborative research environment
📍 Location Highlight
Bayreuth is a charming university town in Germany, offering a peaceful lifestyle, strong research culture, and easy access to major cities like Munich and Nuremberg 🇩🇪
🔗 More Info (Copy Link)
phdscanner.com/opportunities…
🚀 Shape the future of sustainable materials with AI and data-driven innovation!
#PhD #Germany #MaterialsInformatics #ArtificialIntelligence #MachineLearning #Sustainability #Polymers #ComputationalScience #Research #PhDOpportunity
2
7
438
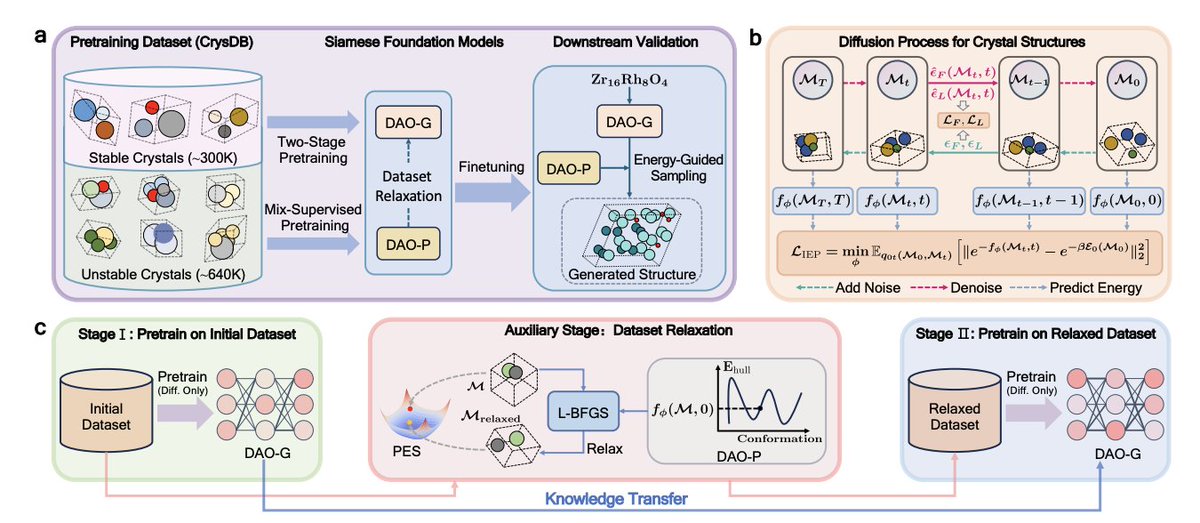
Siamese Foundation Models for Crystal Structure Prediction
1. The paper introduces DAO (Diffusion-based Crystal Omni), a pretrain–finetune framework for crystal structure prediction that couples two “Siamese” foundation models: DAO-G (structure generator) and DAO-P (energy predictor) that actively supports generation via relaxation and energy-guided sampling.
2. A key design choice is to pretrain on both stable and unstable crystals, rather than stable-only corpora. The authors build CrysDB (~940K crystals from Materials Project OQMD, with energy annotations), then deduplicate it to avoid leakage into MP-20 and MPTS-52 benchmark test sets.
3. DAO-G is pretrained directly on the CSP objective (learning p(lattice, coordinates | composition)) using a DiffCSP-style diffusion formulation over lattice and fractional coordinates, aiming to make pretraining tightly aligned with downstream CSP finetuning.
4. DAO-P is pretrained with a mix-supervised objective: (a) self-supervised diffusion losses to learn structure denoising signals, plus (b) a supervised energy objective designed to predict intermediate energies along the diffusion trajectory. They propose an exponential energy loss motivated by Boltzmann-constrained modeling to make intermediate-energy learning feasible without explicit intermediate labels.
5. The generator pretraining uses a two-stage pipeline: Stage I trains DAO-G on the full (stable unstable) CrysDB; then DAO-P relaxes moderately unstable structures (Ehull in (0.08, 0.5] eV/atom) using predicted energy gradients with L-BFGS; Stage II continues pretraining DAO-G on this “relaxed” dataset to reduce bias toward unstable regions.
6. Architecturally, both models share Crysformer, a geometric graph Transformer designed to respect O(3) symmetry and periodic invariance. It combines invariant attention over graph edges (with Fourier features for periodicity), gated residual connections, and separate heads for noise (generation) and energy (prediction).
7. On CSP benchmarks, large-scale pretraining consistently improves results across backbones. With comparable parameter budgets, Crysformer-based models outperform scaled baselines, and swapping diffusion for flow matching within the same architecture further improves match rate (reported best: 74.17% on MP-20 and 42.01% on MPTS-52 in 1-shot).
8. Ablations support the “generator–predictor synergy”: including unstable crystals helps, but adding DAO-P-based relaxation (Stage II) further improves generation quality; and energy-guided sampling increases stability rates of generated structures (e.g., MP-20 stability 85.99% → 87.42%; MPTS-52 73.75% → 75.05%) and can especially help on harder, larger systems.
9. DAO-G is evaluated for polymorphism recovery (multiple structures per composition), showing strong coverage in 2-/3-/4-polymorph cases and demonstrating that the model can generate diverse conformations for the same composition rather than collapsing to a single mode.
10. Real-world validation targets superconductors: after finetuning on SuperCon-derived 3D data, DAO-G predicts structures for unseen superconductors (Cr6Os2, Zr16Rh8O4, Zr16Pd8O4). For Cr6Os2, it reports 100% match rate with extremely low atomic-position error (RMSE 0.0012 over 20-shot), and DFT-computed Ehull closely matches experiment; generation iterations are reported >2000× faster than a DFT-based optimizer per iteration. DAO-P also supports Tc prediction, improved by augmenting missing-structure entries with DAO-G-generated structures.
💻Code: github.com/ManlioWu/DAO
📜Paper: arxiv.org/abs/2503.10471
#CrystalStructurePrediction #MaterialsInformatics #GenerativeModels #DiffusionModels #FlowMatching #FoundationModels #GraphTransformers #Superconductors #ComputationalMaterials #AIforScience
2
15
1,528

How can molecular structures and physicochemical properties be estimated from NMR spectra using QSPR models?
This document introduces a reverse analysis workflow integrating NMR data with QSPR modeling.
👉jeol.com/solutions/applicati…
#NMR #QSPR #MaterialsInformatics #JEOL
1
2
402

Feb 19
The excitement continues to build on Day 01 at ICMG-IV as the focus shifts towards one of the most transformative developments in modern research — large language models in materials science.
Now taking the stage is Prof. Taylor D. Sparks (@taylordsparks), Professor at the University of Utah, USA. His Invited Lecture, “The LLM Revolution in Materials Science: From Data Extraction to Crystal,” is capturing the imagination of the audience.
The hall is abuzz with curiosity as researchers witness how machine intelligence is redefining discovery pipelines in real time. ICMG-IV is proving to be a meeting ground where computation, data science, and materials innovation converge powerfully.
#ICMG #ICMG2026 #ICMGIV #ACCMS #SRMUniversityAP #SRMAP #SRMAmaravati #SRM #SRMUniversity #SRMAPResearch #ResearchAtSRMAP #LargeLanguageModels #AIinScience #MaterialsInformatics #DataDrivenResearch #CrystalEngineering #ComputationalMaterials #ScientificInnovation




2
39

#AI is changing the way new materials are discovered, designed, and brought to life.
In our latest whitepaper, we explore how #MaterialsInformatics is enabling faster, smarter, and more sustainable material discovery. By combining active learning, generative design, and autonomous labs, AI is helping researchers move from insight to #innovation with greater speed and confidence.
Discover how @tech_mahindra is building future‑ready materials research ecosystems through AI‑powered modeling, robust data curation platforms, and strong industry‑academia collaboration, turning advanced research into real‑world impact.
👉 Read more: techmahindra.com/insights/wh…
#ScaleAtSpeed #MakersLab #MachineLearning #MaterialsScience

1
1
142

Jan 27
【明日からnanotech 2026に出展 🔬】
材料探索から物性評価、表面・界面現象の解析、結晶構造探索まで。
原子レベルシミュレーション「Matlantis」を用いて、実験と計算をどのように組み合わせ、材料研究を加速できるのかをご紹介します。
ブースでは、
・計算が研究現場でどのように役立つのか
・どのような物性を具体的に計算できるのか、その活用例
・企業/大学での導入による変化や活用のインタビュー
などを、デモやミニセミナーを交えてご覧いただけます。
材料開発のスピード向上や、試行錯誤の削減に関心のある方は、ぜひお立ち寄りください。
📅 1/28(水)– 30(金)10:00–17:00
📍 東京ビッグサイト West 1・3ホール&会議棟
🎪 小間番号:3W-C22
#nanotech #材料研究 #計算化学 #MI #MaterialsInformatics #Matlantis #材料開発

1
10
691

10 Dec 2025
Next-gen battery breakthrough! 🔋 We're using AI to simulate the atomic structure of the SEI layer—over 120,000 atoms at a time. This leap in materials science accelerates development from years to months. The future of energy is predictive, not trial-and-error.
blog-en.fltech.dev/entry/202…
#Batteries #AI #MaterialsInformatics #Innovation

ALT Next generation battery
1
5
114

5 Dec 2025
Meet Dr Rohit Batra, Assistant Professor in the Department of Metallurgical and Materials Engineering at IIT Madras, working at the forefront of materials informatics. His research uses AI/ML to design new materials and optimise processing techniques, driving innovation toward a more sustainable future. He is especially excited about the rise of self-driving labs, where AI and robots accelerate materials discovery.
As a mentor, Dr Batra believes in helping students reach their highest potential through motivation, constructive feedback, and an inclusive work environment that encourages equal contribution.
Balancing his roles with passion, he introduces industry-relevant
courses, pursues cutting-edge research, and contributes to
administrative activities where he can make a meaningful impact.
And yes—his classes are known for student-run buzzer quizzes, and his group meetings sometimes turn into mini concerts!
Learn more about his work here: mme.iitm.ac.in/innerfaculty.….
Find them on LinkedIn:
linkedin.com/company/materia…
@iitmadras
#IITMadras #FacultySpotlight #MaterialsInformatics #ResearchAtIITM #AIforScience
4
785
21 Nov 2025
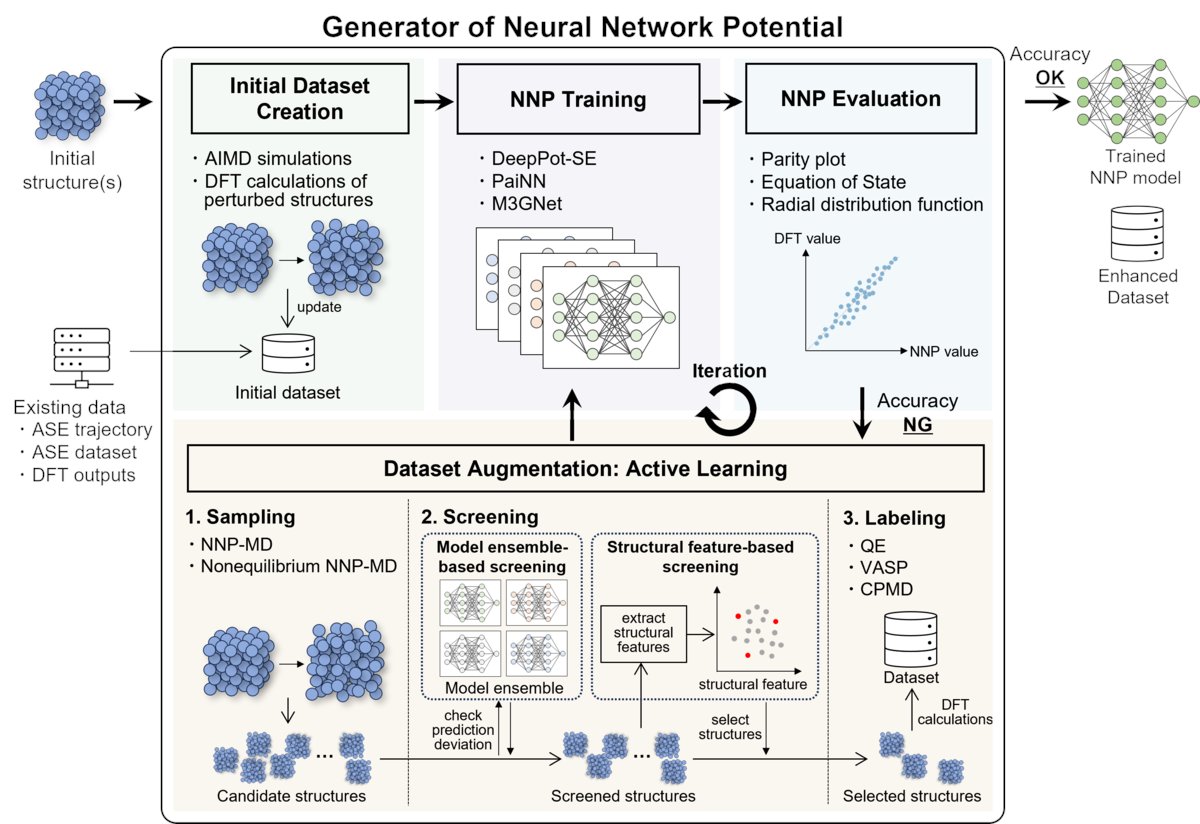
🔬✨【Materials Informatics特集 #14】
今回は、分子動力学シミュレーション向けニューラルネットワーク力場の作成するツールGeNNIP4MDを活用した、日本製鉄様によるニッケル合金の水素脆化の解析事例の論文内容をご紹介します。
水素脆化を根本的に解決し、より安全で信頼性の高い水素関連インフラを構築するためには、材料内部における水素の挙動、特に「拡散」と「溶解」のメカニズムを原子レベルで詳細に理解することが不可欠です。
また、複雑な合金系における水素挙動の解明には、従来のシミュレーション手法では限界がありました。
このような技術課題を克服するために、私たちはGeNNIP4MDを用いた新しいアプローチを提案しました。本研究では、Ni-Mn-H三元系という複雑な系において、最小限のDFT計算コストで、広範な原子配置を正確に記述できる高精度なNNPを効率的に構築することに成功しました。このNNPを用いることで、DFTに匹敵する精度を保ちながら、従来のMDシミュレーションでは不可能だった大規模かつ長時間の水素拡散シミュレーションが可能となりました。
👉 詳細はこちら:blog.fltech.dev/entry/2025/0…
#MaterialsInformatics #GeNNIP4MD
ALT GeNNIP4MDの概要
4
373

14 Nov 2025
🔬 New in JMI: Stacked machine learning for accurate and interpretable prediction of MXenes’ work function
Open access👉oaepublish.com/articles/jmi.…
#MXene #MachineLearning #MaterialsInformatics

3
64
22 Oct 2025
🔬✨【Materials Informatics特集 13】
今回は、分子動力学シミュレーション向けニューラルネットワーク力場を作成するツールGeNNIP4MDの「知識蒸留」という新機能を活用し、半導体製造の重要プロセスである半導体表面のシリコン酸化膜(シリカ, SiO2)をフッ酸によりウェットエッチングする過程を、固液界面モデルの分子動力学 (Molecular Dynamics, MD) シミュレーションで再現した事例を紹介します。
ウェットエッチングのような液中での化学反応は、実験的にその過程を原子レベルで直接観察することが極めて困難です。GeNNIP4MDの知識蒸留では、大量のデータで訓練された事前学習済みの機械学習力場を教師モデルとして、その知識を軽量な生徒モデルに効率よく移すことができます。また、従来は開発が困難であった「化学反応を高精度に記述でき、かつ大規模・長時間のシミュレーションが可能な機械学習力場」を、少ない計算コストで自動的に構築することが可能になります。
👉 詳細はこちら:blog.fltech.dev/entry/2025/0…
#MaterialsInformatics #GeNNIP4MD

4
229
14 Oct 2025
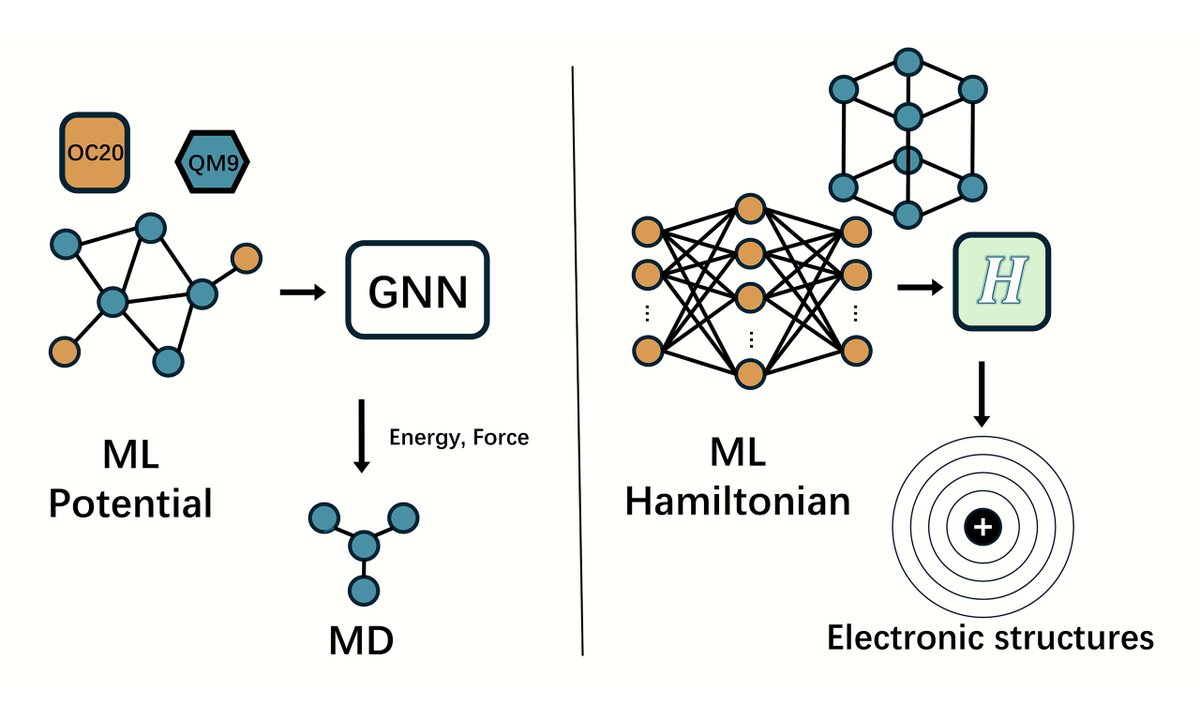
🔥 Must-read! Prof. Lei Shen’s team (NUS) reviews ML interatomic potentials & Hamiltonians—key advances, challenges, and trends driving AI4Science & materials discovery.
👉 Dive in: oaepublish.com/articles/jmi.…
#AI4Science #MachineLearning #MaterialsInformatics
5
223