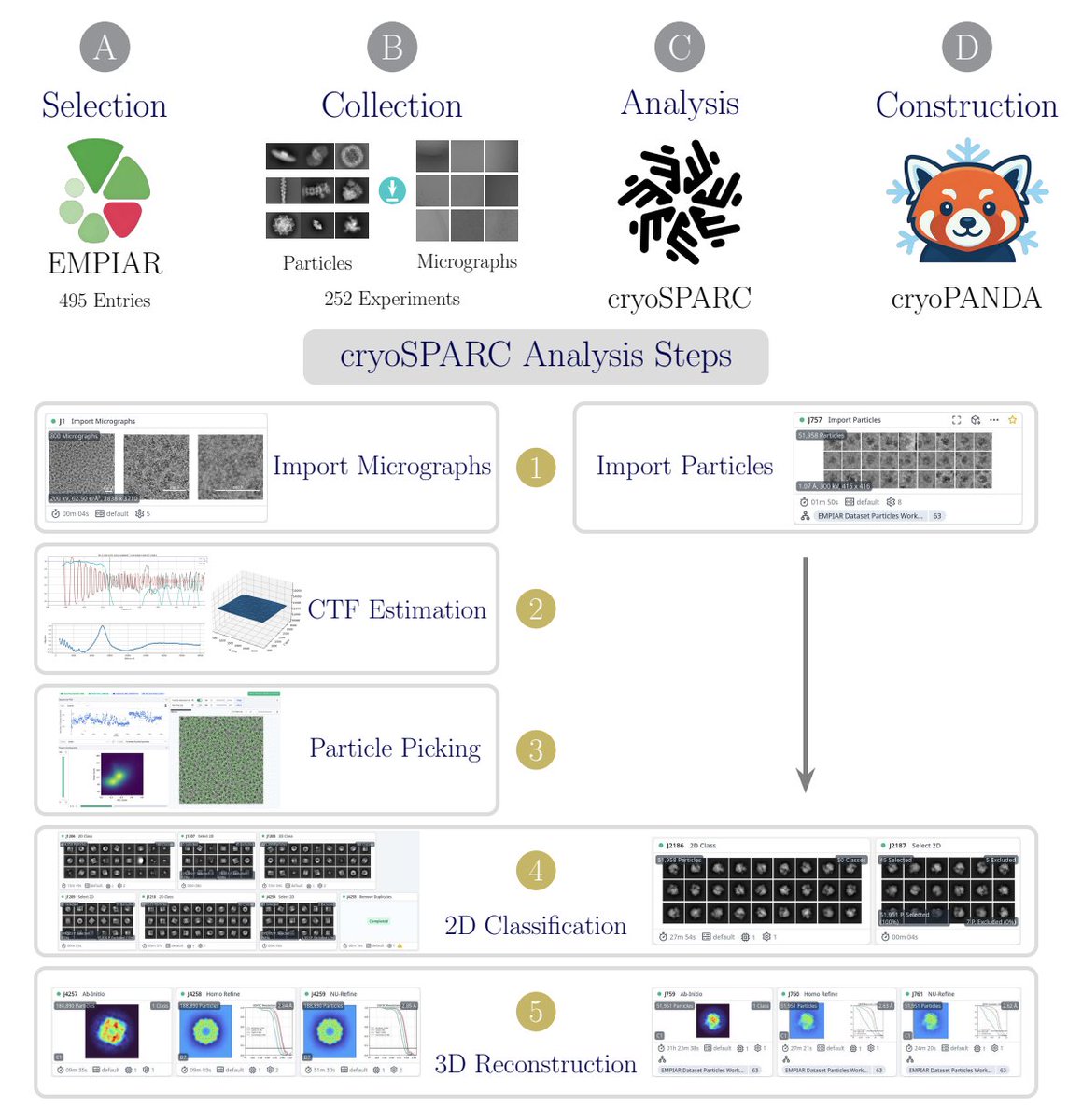
A 37-million-particle dataset from over 250 experiments to accelerate data-driven cryo-EM analysis
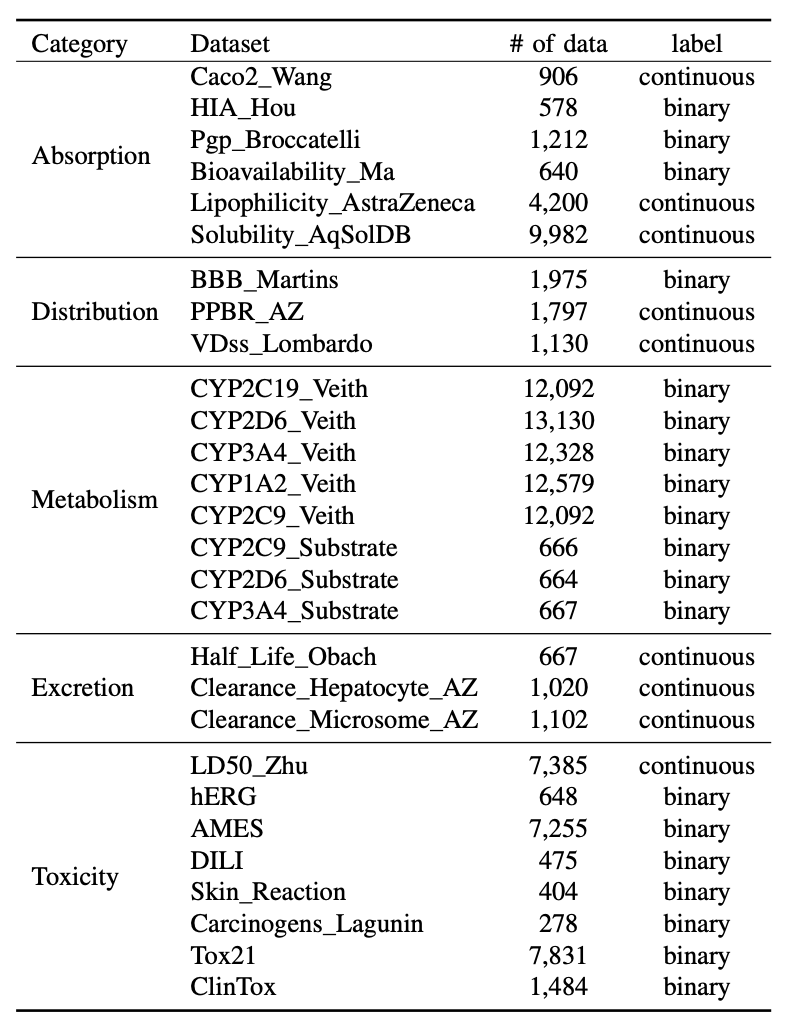
1. The paper introduces cryoPANDA (cryo-EM Particles ANnotated DAtaset): 37,623,123 curated experimental particle images from 252 cryo-EM experiments, designed to remove the main bottleneck for particle-level foundation models in cryo-EM: lack of large, diverse, richly annotated real data.
2. Scale and diversity are key: cryoPANDA spans 16 function-based protein classes and broad molecular-weight ranges (mean ~600 kDa; min 21 kDa; max 200,000 kDa), aiming to support models that generalize across targets and imaging conditions rather than being retrained per experiment.
3. Rich per-particle annotations go far beyond picking coordinates, covering acquisition parameters (e.g., voltage, dose, Cs), CTF estimates (defocus U/V, astigmatism angle), 2D classification statistics (class, alignment resolution, ESS, ECA), and 3D reconstruction metadata (Euler angles, translations, alignment error), plus links to EMDB maps and (when available) PDB models.
4. Dataset construction is not a simple scrape: the authors examined 495 EMPIAR entries, used sequence similarity (>30%) to cluster entries and reduce redundancy, then selected up to four representatives per cluster with manual curation for data quality and documentation, yielding 252 final experiments (mostly EMPIAR 5 in-house).
5. A standardized cryoSPARC v4.6 processing pipeline is used to curate particles and attempt reconstructions: CTF estimation (when starting from micrographs), picking (blob picker or author coordinates), multiple rounds of 2D classification/selection with recovery of mistakenly rejected classes, duplicate removal using estimated particle diameter, and typical ab initio refinement steps for 3D maps.
6. Reconstruction quality is validated against published EMDB maps (for cases with reported reconstructions): among 214 experiments with cryoPANDA reconstructions, 75 (35%) achieve better reported resolution than the published map and 139 (65%) are worse; differences are often explained by cryoPANDA using smaller particle subsets, with results becoming broadly comparable when particle fractions match.
7. A major contribution is demonstrating foundation-model readiness: the authors train a DINOv2 ViT-L/16 model from scratch on ~32M particles (215 experiments) and test generalization on 37 held-out experiments (~5M particles), using an experiment-level split to avoid leakage across near-identical acquisition settings or targets.
8. Without task-specific fine-tuning, the pretrained model yields micrograph-level representations that separate particle regions from background via sliding-window feature extraction and PCA-to-RGB visualization, despite the model being trained only on cropped particle images (not full micrographs).
9. The paper also shows a fully unsupervised particle-picking pipeline built on frozen DINOv2 features, evaluated on held-out EMPIAR-10017 with Henderson’s manual annotations: 91.5% recall, 45.5% precision (F1 60.8%). After downstream cryoSPARC cleanup, the picked particles support a 3D reconstruction at 4.38 Å, close to the published 4.20 Å and the cryoPANDA pipeline’s 4.29 Å for the same dataset.
10. Using cryoPANDA’s metadata, linear probes on frozen DINOv2 features can predict multiple particle properties (symmetry, pixel size, molecular weight, max diameter, EMDB resolution, defocus). Cross-experiment performance drops vs in-distribution, and the authors quantify that part of this gap comes from acquisition-parameter entanglement; regressing out acquisition parameters improves OOD accuracy across tasks, illustrating how the dataset enables mechanistic analysis of generalization failures.
💻Code:
github.com/azamanos/cryoPAND…
📜Paper:
biorxiv.org/content/10.64898…
#cryoEM #StructuralBiology #DeepLearning #FoundationModels #SelfSupervisedLearning #Datasets #Bioinformatics #ComputationalBiology #EMPIAR #EMDB