
Jun 12
This work is part of the foundation for my Polarized Hypergraph Spectral Program and reliable numerics for geometric/simulation projects.
Everything is open: 12-page note LaTeX source large-scale GPU logs minimal examples.
If you work with equivariant spectral methods, TDA, or geometric topology — I’d love to hear your feedback, reproductions, or similar stories you’ve encountered.
DOI: doi.org/10.5281/zenodo.20671…
Repo: github.com/franklino79-TPCD/…
6/6
#HodgeLaplacian #SpectralMethods #TopologicalDataAnalysis #GeometricTopology #QuotientComplex #NumericalLinearAlgebra #OpenScience
21

Jun 8
📢 Call for Papers 📢
Special Issue on "15th Anniversary of Axioms: Geometry and Topology"
Guest Editor: Dr. Emil Saucan
🔗 Find out more and submit your paper now: brnw.ch/21x3anz
#topologicaldataanalysis #discretecurvatures #curvatureflows #CallForPapers

1
21

Apr 30

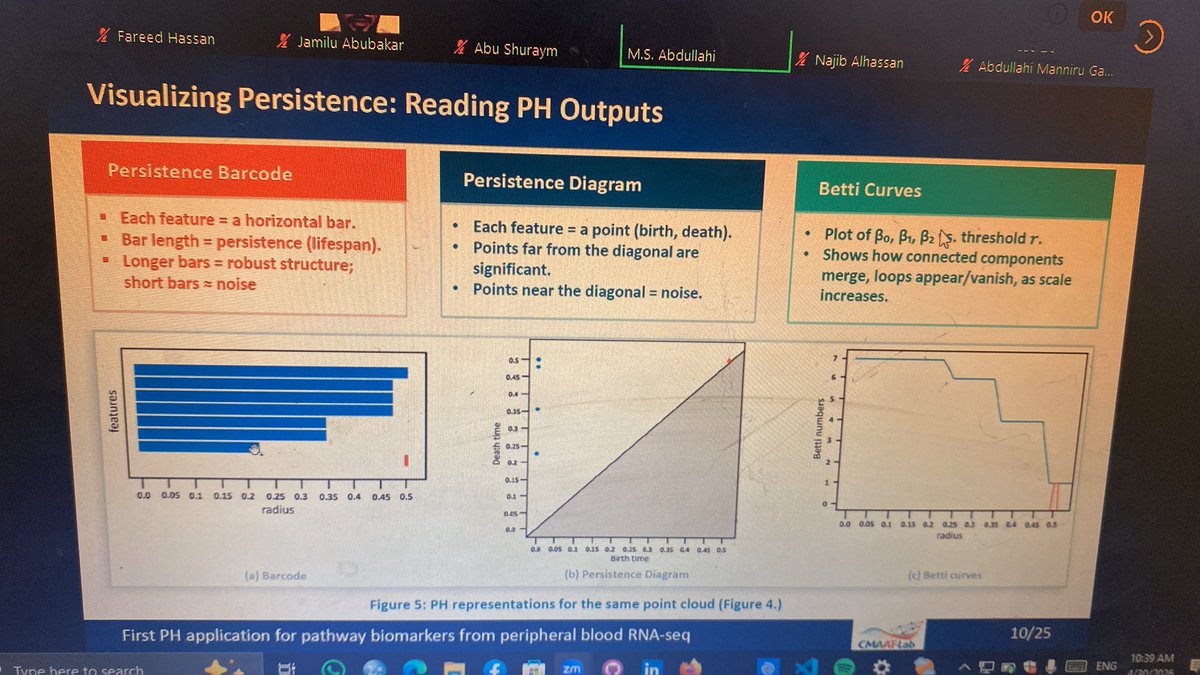
2
2
204
Protein Language Models Encode Evolutionary Grammar but Conflate Topological and Thermodynamic Phases
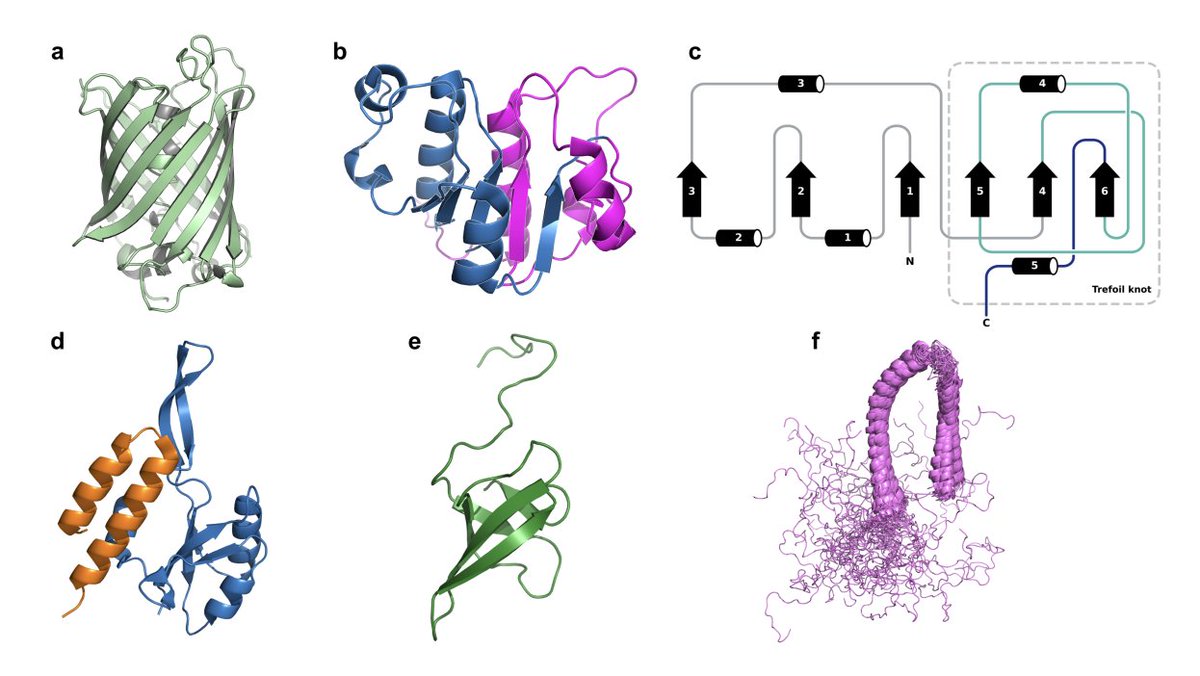
1. Wang et al. probe what a sequence-only protein language model (ESM-2 3B) actually encodes by stress-testing it on key “Anfinsen exceptions”: intrinsically disordered proteins (IDPs), fold-switching proteins, and knotted proteins—cases where sequence-to-structure is not a single static mapping.
2. Core result: ESM-2 largely discards microscopic 3D backbone geometry during embedding formation, and instead builds a macroscopic “sequence grammar manifold” shaped by evolutionary statistics and physicochemical composition—useful for separating biological from unphysical sequences, but weak for topology/phase distinctions.
3. To test microscopic geometric awareness, the study uses Hasimoto integrability error E[n], a differential-geometric order parameter tied to backbone twisting/folding symmetry breaking. Residue-level correlations between embedding distances and E[n] are negligible (overall Spearman ρ ≈ 0.105; R² ≈ 0.015), arguing against atomic-detail geometry being represented in the latent space.
4. Global latent structure: PCA of 11,068 proteins reveals a horseshoe-shaped manifold. Random sequences form a clearly isolated cluster (Silhouette ≈ 0.344 in 50D PCA; per-sample mean ≈ 0.566), indicating strong sensitivity to “evolutionary plausibility” of sequences.
5. The main manifold axes map to composition more than geometry: PC2 correlates with hydropathy (GRAVY), pI, and especially aromaticity (ρ ≈ 0.364), creating hydrophilic–hydrophobic gradients. SCOP classes show partial ordering, consistent with statistical secondary-structure preferences rather than explicit coordinate encoding.
6. Key limitation: “topological aliasing.” IDPs, knotted proteins, and fold-switching proteins are not separable in ESM-2 space (negative Silhouette means: knotted ≈ −0.151, fold-switching ≈ −0.108, IDP ≈ −0.057). The model conflates physically distinct topological/thermodynamic regimes when sequence statistics overlap.
7. A region-replacement control argues the conflation is intrinsic, not just mean-pooling “dilution.” Replacing annotated distinctive regions (disordered segments / knot regions / fold-switching interface units) with matched ASTRAL95 regions barely changes Silhouette scores (shifts ~0.0–0.6%), implying the limitation is not localized to a removable motif.
8. Density behavior in latent space inverts physical entropy: using KDE on UMAP-2D, IDPs (physically high conformational entropy) occupy the densest latent regions (IDP density ~1.36× ASTRAL95 baseline), interpreted as low evolutionary sequence entropy being compressed into tight manifold neighborhoods.
9. Mechanistic explanation via topology “gauge” geometry: persistent homology separates random vs biological classes at a macroscopic level (large Wasserstein-2 distances), but holonomy-defect analysis shows class-invariant local curvature (“geometric turbulence”; tiny effect sizes η²), explaining why local neighborhoods fail to resolve fine topological/thermodynamic phases.
10. Structure-aware control: SaProt (sequence Foldseek 3Di tokens) partially reduces aliasing for static anomalies like knots (Silhouette knotted: −0.106 → 0.008; 8% → 56% positive), but still cannot separate alternative fold states in fold-switching proteins (conf1 vs conf2 Silhouette ≈ −0.002), suggesting static structural tokens help topology but not multi-state thermodynamic phase behavior.
💻Code: github.com/wyqmath/ESM-Laten…
📜Paper: biorxiv.org/content/10.64898…
#ProteinLanguageModels #ESM2 #ComputationalBiology #ProteinFolding #IntrinsicDisorder #FoldSwitching #ProteinKnots #TopologicalDataAnalysis #RepresentationLearning #Biophysics
6
55
3,663

Feb 13
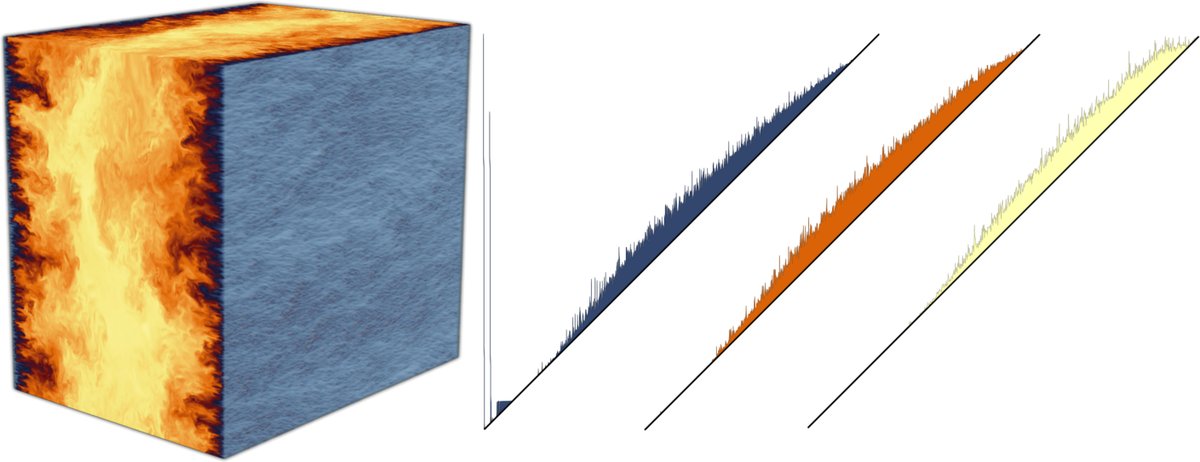
😍Computing the persistence diagrams of a turbulent flow dataset of 6 billion vertices?
#TTK now does it in less than 3 minutes on @Sorbonne_Univ_ distributed supercomputer!
Paper: arxiv.org/abs/2505.21266
Example: topology-tool-kit.github.io/…
#TopologicalDataAnalysis #HPC #DataScience
2
4
177
😍 Check out our latest entry in the #TopologyToolKit Online Example Database!
🧑🎓 Today, learn how to robustly track topological features in time-varying data, in just 42 lines of #Python 👇
topology-tool-kit.github.io/…
#Visualization #TopologicalDataAnalysis #DataScience

7
305

Jan 28
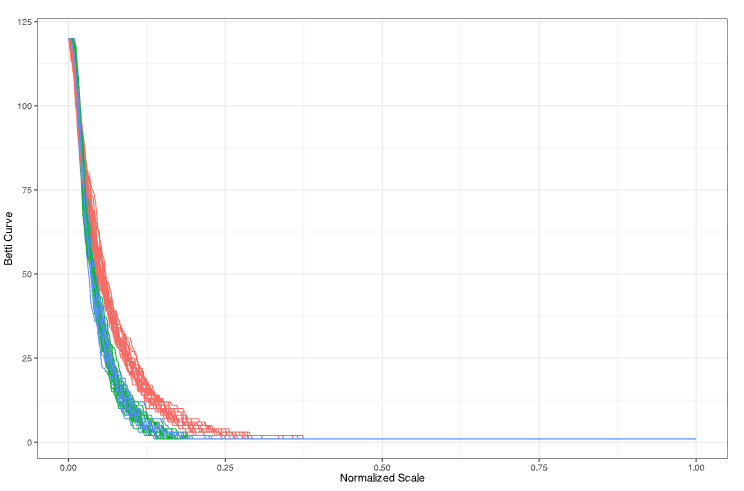
ISC project update: {inphr} brings statistical inference to persistence diagrams in R (permutation tests diagram distances; plus curve-based summaries via {TDAvec} to localize differences).
Read: r-consortium.org/posts/stati…
#rstats #TopologicalDataAnalysis #OpenSource
3
477
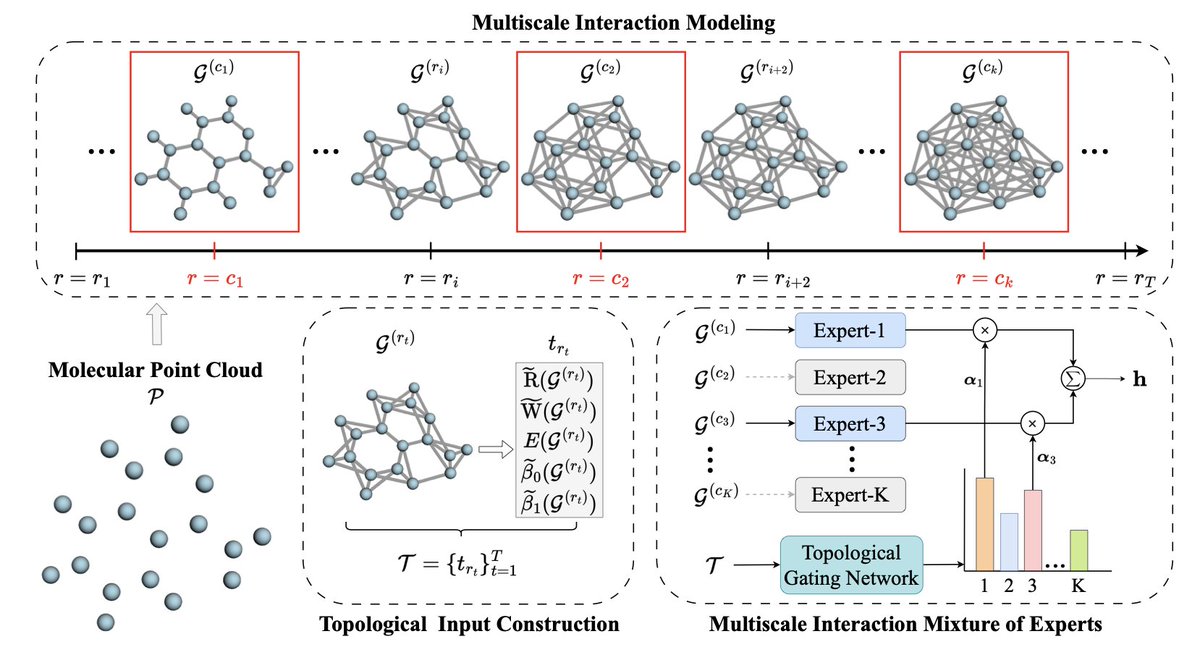
Topology-Aware Multiscale Mixture of Experts for Efficient Molecular Property Prediction
1. A new framework, MI-MoE, has been proposed to enhance the prediction of molecular properties by integrating a multiscale mixture of experts with topology-aware gating. This approach addresses the limitation of fixed interaction cutoffs in traditional 3D molecular graph neural networks, enabling more flexible and adaptive modeling of molecular interactions.
2. The core innovation of MI-MoE is its ability to capture short-, mid-, and long-range interactions through a set of experts defined by different distance cutoffs. This multiscale approach allows the model to adaptively select and combine interaction scales based on the specific characteristics of each molecule, leading to improved accuracy in property prediction.
3. The gating mechanism in MI-MoE leverages topological descriptors derived from persistent homology and filtration-based features. These descriptors summarize how molecular connectivity evolves across different radii, providing a robust signal for adaptive routing and expert selection. This integration of topological information is a novel aspect that enhances the model's ability to handle diverse molecular structures.
4. Extensive experiments demonstrate that MI-MoE consistently outperforms single-scale models and other state-of-the-art baselines across various molecular and polymer property prediction benchmarks. The framework shows significant improvements in both regression and classification tasks, highlighting its versatility and effectiveness.
5. MI-MoE is designed as a plug-and-play module, making it easily integrable with different 3D GNN architectures. This flexibility allows researchers to enhance existing models without extensive modifications, facilitating broader adoption and further development in the field of molecular property prediction.
📜Paper: arxiv.org/abs/2601.12637v1
#MolecularPropertyPrediction #GraphNeuralNetworks #TopologicalDataAnalysis #MachineLearning #ComputationalChemistry
9
19
1,725
19 Dec 2025
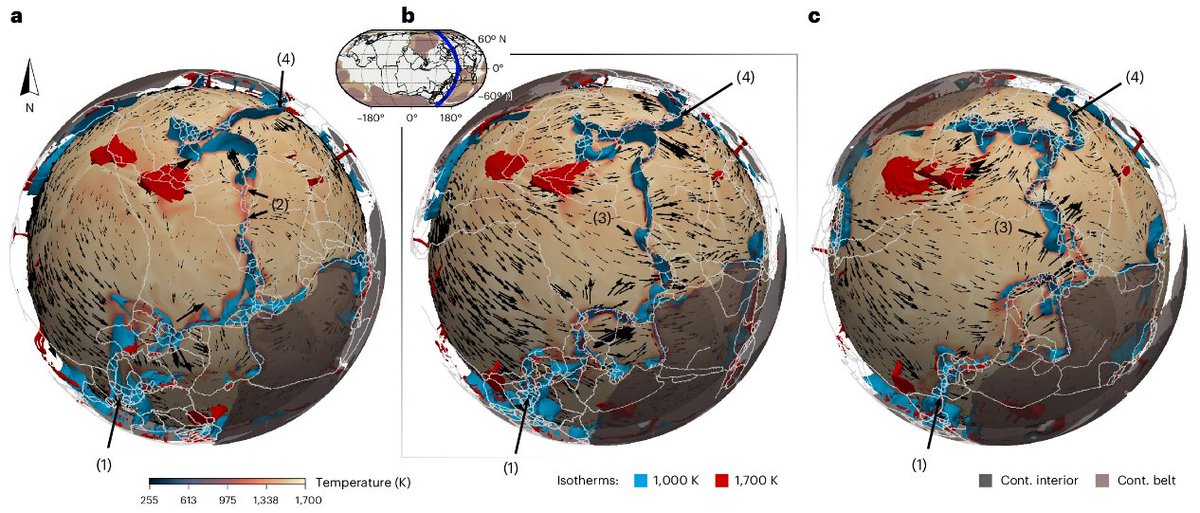
🚨 Paper alert!
Checkout our @NatureGeosci paper on the topological analysis of geodynamics data, where we track topological features through time to characterize tectonic plate displacements 🤔
👉 nature.com/articles/s41561-0…
#TopologicalDataAnalysis #Geodynamics
6
14
2,432

11 Dec 2025
In my opinion, cosmology’s next frontier is structure topology ML 🌠
#Cosmology #AI4Science #TopologicalDataAnalysis #NonGaussianity
1
1
6
1,571
5 Dec 2025
🚨 Paper alert!
Checkout our latest #ieeevis paper on the stability analysis of h-bonds in collections of quantum chemistry datasets. H-bonds are surprisingly robust to molecular vibrations and proton tunneling! 🤔
👉 arxiv.org/abs/2504.03205
#TopologicalDataAnalysis #QTAIM

4
277

4 Dec 2025
新しいプレプリントを公開しました(2025年12月3日):
「LLM埋め込み空間における持続的トポロジー構造:
幾何学的分析から制御可能性へ」
(Meaning Unification Framework の Tier-I)
LLM の埋め込み空間に、アーキテクチャに依存しない
安定な persistent H₁ サイクルが存在しており、
強いノイズや層方向の摂動にも崩れないことを示しました。
これらのH₁サイクルは、
LLMをより堅牢にステアリング/アラインメントするための
“トポロジー的に保護された部分空間”として機能し得ることを提案しています。
オープンアクセス(再現コードつき):
zenodo.org/records/17785728
TDA / トポロジカルDLのコミュニティの皆さまから、
ご意見やフィードバックをいただけると嬉しいです。
#TDA #TopologicalDataAnalysis #TopologicalDeepLearning
#PersistentHomology #LLM #AIalignment

New preprint (Dec 3, 2025):
Persistent Topological Structures in LLM Embedding Spaces:
From Geometric Analysis to Controllability
(Tier-I in the Meaning Unification Framework)
We find architecture-agnostic persistent H₁ cycles in LLM embedding spaces that survive strong noise & layer perturbations — suggesting they can act as topologically protected subspaces for robust steering/alignment.
Open access full repro code:
zenodo.org/records/17785728
Would love thoughts from the TDA & Topological DL community — tagging a few people whose work heavily inspired this:
@ninamiolane @HajijMustafa @mathildepapillo @tolga_birdal
@svpino @LidaKanari @AlicePatania
#TDA #TopologicalDataAnalysis #TopologicalDeepLearning #PersistentHomology #LLM #AIalignment
2
263

New preprint (Dec 3, 2025):
Persistent Topological Structures in LLM Embedding Spaces:
From Geometric Analysis to Controllability
(Tier-I in the Meaning Unification Framework)
We find architecture-agnostic persistent H₁ cycles in LLM embedding spaces that survive strong noise & layer perturbations — suggesting they can act as topologically protected subspaces for robust steering/alignment.
Open access full repro code:
zenodo.org/records/17785728
Would love thoughts from the TDA & Topological DL community — tagging a few people whose work heavily inspired this:
@ninamiolane @HajijMustafa @mathildepapillo @tolga_birdal
@svpino @LidaKanari @AlicePatania
#TDA #TopologicalDataAnalysis #TopologicalDeepLearning #PersistentHomology #LLM #AIalignment
1
2
328
28 Nov 2025
🚨 Paper alert!
Checkout our latest #topoinvis paper on topology-aware neural interpolation of time-varying data.
Reconstruct missing time steps with accurate geometry and topology!
👉 arxiv.org/abs/2508.17995
#TopologicalDataAnalysis #Visualization #PersistenceOptimization

2
153
31 Oct 2025
🥳 The @ErcTori team ❤️ @ieeevis!
This year we'll be presenting 1 full paper (honorable mention award!) and 1 #TopoInVis paper:
arxiv.org/abs/2504.03205
arxiv.org/abs/2508.17995
We'll also run a #TopologyToolKit tutorial on Monday! Join us, won't you? #TopologicalDataAnalysis #viz


2
146

28 Aug 2025
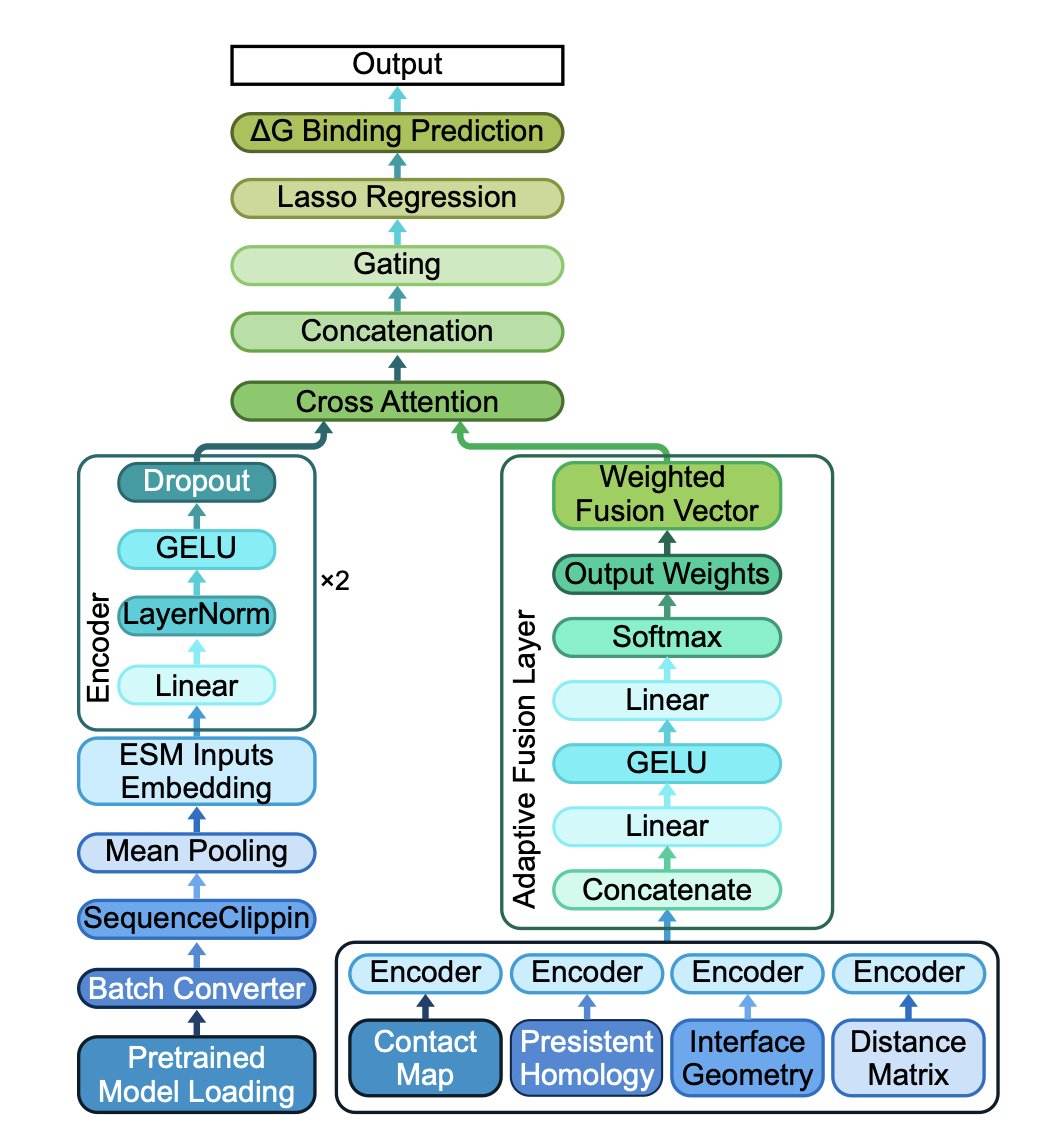
TOPOBIND: MULTI-MODAL PREDICTION OF ANTIBODY-ANTIGEN BINDING FREE ENERGY VIA SEQUENCE EMBEDDINGS AND STRUCTURAL TOPOLOGY
1. A novel framework called TopoBind integrates sequence-based representations from pre-trained protein language models (ESM-2) with a set of topological features to predict antibody-antigen binding free energy, achieving state-of-the-art accuracy in binding free energy prediction.
2. TopoBind extracts contact map metrics, interface geometry descriptors, distance map statistics, and persistent homology invariants to capture both local and global structural organization within individual proteins and across the antibody-antigen interface.
3. The model employs a cross-attention mechanism to fuse diverse modalities effectively, enhancing the prediction of binding free energy. It also uses an adaptive feature fusion mechanism to dynamically weight different topological submodules, improving model generalization.
4. Experiments on a curated dataset of 303 antibody-antigen complexes show that TopoBind consistently outperforms sequence-only and conventional structural models in both regression and classification settings.
5. Ablation studies demonstrate the importance of each architectural component, including the adaptive fusion module and sparse linear modeling, in enhancing the model's generalization ability.
6. The study also explores the sensitivity of topological parameters, finding that the model performs best with specific settings for the interface contact distance threshold and the number of top-k persistent homology lifetimes retained.
📜Paper: arxiv.org/abs/2508.19632
#TopoBind #AntibodyAntigenBinding #ProteinLanguageModels #TopologicalDataAnalysis #StructuralBioinformatics #CrossAttention #ProteinRepresentationLearning #MolecularMachineLearning
2
22
1,402
30 Jul 2025
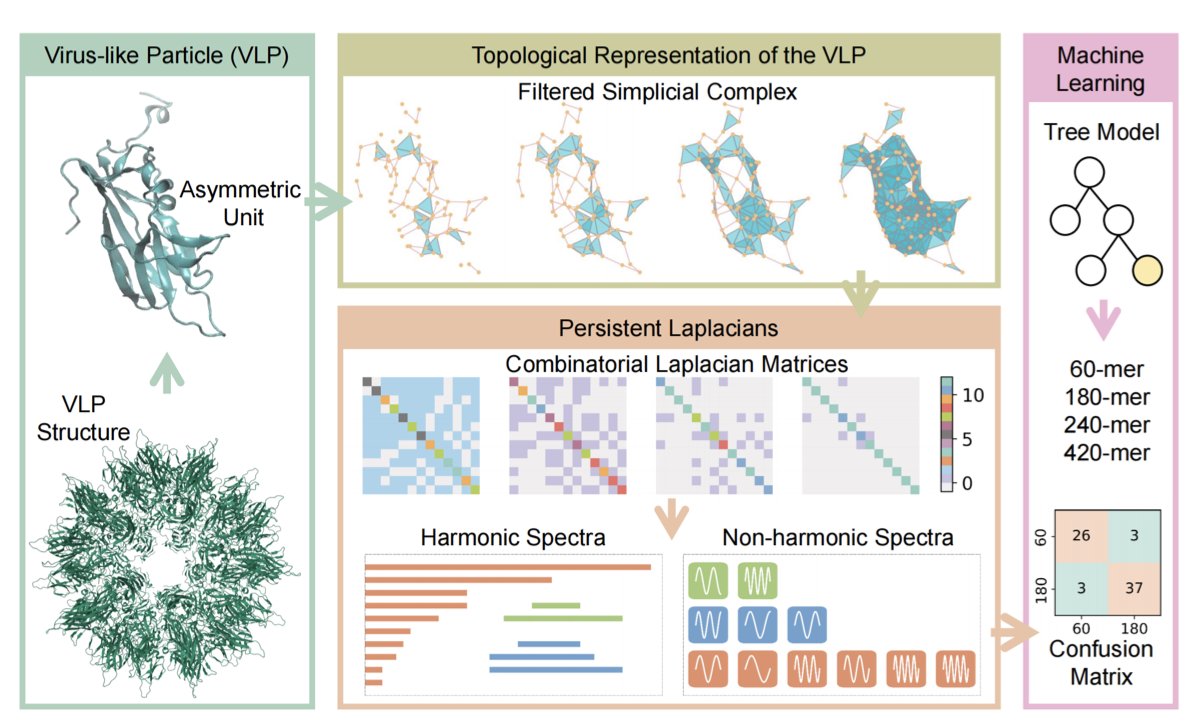
Machine-Learning Prediction of Virus-like Particle Stoichiometry and Stability using Persistent Topological Laplacians
1. A novel machine learning model leveraging persistent Laplacians has been introduced to predict the stoichiometry and stability of virus-like particles (VLPs). This approach captures intricate topological and geometric features of VLP structures, outperforming existing methods on the VLP200 dataset.
2. The study expands the dataset to VLP706, comprising 706 samples with diverse stoichiometries (60-mer, 180-mer, 240-mer, and 420-mer). The model maintains strong predictive accuracy, achieving an AUC of 0.956 and accuracy of 0.858 in 10-fold cross-validation.
3. The model uses filtered simplicial complexes (Vietoris-Rips and Alpha complexes) to represent VLP structures, extracting topological features via persistent Laplacians. These features are then fed into a gradient boosting tree algorithm for classification.
4. Stability analysis through random sequence perturbations reveals that 60-mers and 180-mers exhibit greater stability than 240-mers and 420-mers. This finding suggests that 60-mer and 180-mer VLPs are more robust candidates for vaccine and drug design.
5. The persistent Laplacian approach captures both harmonic (topological) and non-harmonic (geometric) spectra, providing a comprehensive representation of VLP structures. This method has shown superior performance in other biomolecular applications, such as predicting SARS-CoV-2 variants.
📜Paper: arxiv.org/abs/2507.21417v1
#MachineLearning #VirusLikeParticles #TopologicalDataAnalysis #VaccineDesign #DrugDelivery
2
611
30 Jul 2025
Machine-Learning Prediction of Virus-like Particle Stoichiometry and Stability using Persistent Topological Laplacians
1. A novel machine learning model leveraging persistent Laplacians has been introduced to predict the stoichiometry and stability of virus-like particles (VLPs). This approach captures intricate topological and geometric features of VLP structures, outperforming existing methods on the VLP200 dataset.
2. The study expands the dataset to VLP706, comprising 706 samples with diverse stoichiometries (60-mer, 180-mer, 240-mer, and 420-mer). The model maintains strong predictive accuracy, achieving an AUC of 0.956 and accuracy of 0.858 in 10-fold cross-validation.
3. The model uses filtered simplicial complexes (Vietoris-Rips and Alpha complexes) to represent VLP structures, extracting topological features via persistent Laplacians. These features are then fed into a gradient boosting tree algorithm for classification.
4. Stability analysis through random sequence perturbations reveals that 60-mers and 180-mers exhibit greater stability than 240-mers and 420-mers. This finding suggests that 60-mer and 180-mer VLPs are more robust candidates for vaccine and drug design.
5. The persistent Laplacian approach captures both harmonic (topological) and non-harmonic (geometric) spectra, providing a comprehensive representation of VLP structures. This method has shown superior performance in other biomolecular applications, such as predicting SARS-CoV-2 variants.
📜Paper: arxiv.org/abs/2507.21417v1
#MachineLearning #VirusLikeParticles #TopologicalDataAnalysis #VaccineDesign #DrugDelivery

2
3
602
25 Jul 2025
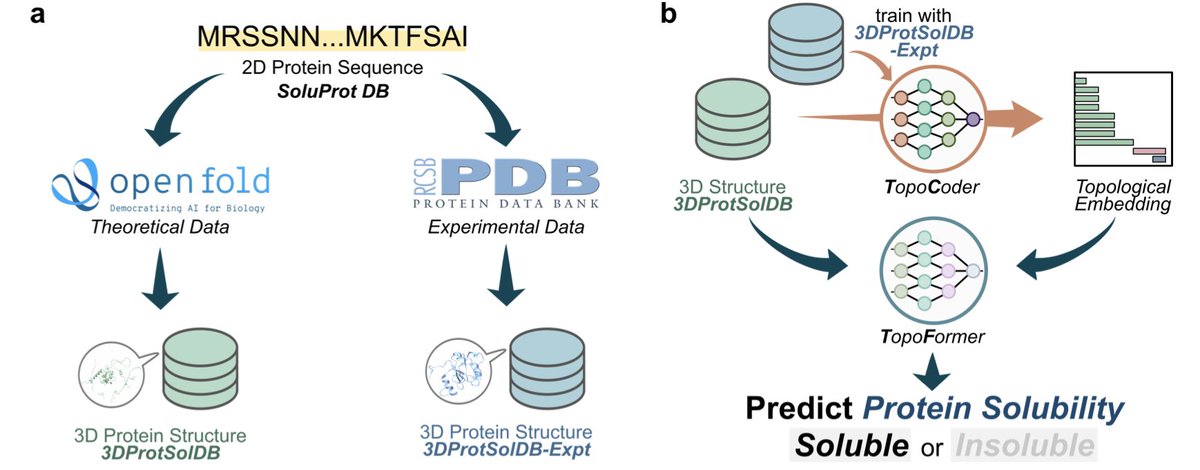
Combining Geometry and Topology for Accurate Protein Solubility Prediction
1. A novel approach to predicting protein solubility using a combination of geometry and topology has been introduced in this study. The model, named TopoFormer, integrates topological embeddings with geometric representations to achieve state-of-the-art accuracy in predicting binary protein solubility from predicted 3D structures.
2. The TopoFormer model leverages a unique combination of equivariant neural networks and topological data analysis to address the challenges posed by the variability in predicted protein structures. This integration allows the model to maintain rotational equivariance while capturing key structural insights, making it robust to input noise.
3. The study demonstrates the creation of two new 3D structural databases, 3DProtSolDB and 3DProtSolDB-Expt, which were used to train the TopoCoder and TopoFormer models. These databases provide a rich source of data for training and validating the models, ensuring their accuracy and generalizability.
4. The TopoCoder model, developed to generate topological embeddings, shows significant resistance to input noise compared to traditional methods. This robustness is crucial for accurately predicting solubility from predicted protein structures, which may contain regions of low confidence.
5. The trained TopoFormer model achieves an accuracy of 81.4% on a held-out test set, outperforming previous models. Additionally, the model's interpretability is highlighted through its ability to identify key residues influencing solubility, which can guide the rational design of soluble protein mutants.
6. The study validates the model's predictions using molecular dynamics simulations, showing a strong correlation between the model's attention weights and water survival probabilities. This correlation underscores the model's utility in capturing physical insights into protein-solvent interactions.
7. The TopoFormer model's ability to predict solubility for carbonic anhydrase mutants demonstrates its potential for practical applications in industrial protein production. The model's predictions align well with experimental data, highlighting its value in guiding protein design.
📜Paper: doi.org/10.26434/chemrxiv-20…
#ProteinSolubility #MachineLearning #TopologicalDataAnalysis #EquivariantNeuralNetworks #ProteinStructurePrediction

3
664
25 Jul 2025
Combining Geometry and Topology for Accurate Protein Solubility Prediction
1. A novel approach to predicting protein solubility using a combination of geometry and topology has been introduced in this study. The model, named TopoFormer, integrates topological embeddings with geometric representations to achieve state-of-the-art accuracy in predicting binary protein solubility from predicted 3D structures.
2. The TopoFormer model leverages a unique combination of equivariant neural networks and topological data analysis to address the challenges posed by the variability in predicted protein structures. This integration allows the model to maintain rotational equivariance while capturing key structural insights, making it robust to input noise.
3. The study demonstrates the creation of two new 3D structural databases, 3DProtSolDB and 3DProtSolDB-Expt, which were used to train the TopoCoder and TopoFormer models. These databases provide a rich source of data for training and validating the models, ensuring their accuracy and generalizability.
4. The TopoCoder model, developed to generate topological embeddings, shows significant resistance to input noise compared to traditional methods. This robustness is crucial for accurately predicting solubility from predicted protein structures, which may contain regions of low confidence.
5. The trained TopoFormer model achieves an accuracy of 81.4% on a held-out test set, outperforming previous models. Additionally, the model's interpretability is highlighted through its ability to identify key residues influencing solubility, which can guide the rational design of soluble protein mutants.
6. The study validates the model's predictions using molecular dynamics simulations, showing a strong correlation between the model's attention weights and water survival probabilities. This correlation underscores the model's utility in capturing physical insights into protein-solvent interactions.
7. The TopoFormer model's ability to predict solubility for carbonic anhydrase mutants demonstrates its potential for practical applications in industrial protein production. The model's predictions align well with experimental data, highlighting its value in guiding protein design.
📜Paper: doi.org/10.26434/chemrxiv-20…
#ProteinSolubility #MachineLearning #TopologicalDataAnalysis #EquivariantNeuralNetworks #ProteinStructurePrediction
4
728