
Fresh off the press! @Crystals_MDPI
Our study on novel organoplatinum(IV) 6,6'-dimethyl complex shows #DNAbinding and #anticancer activity against colon and ovarian carcinomas
mdpi.com/3839122
@nyitcomar
@nyitcomdo
@nyit
@MDPIOpenAccess
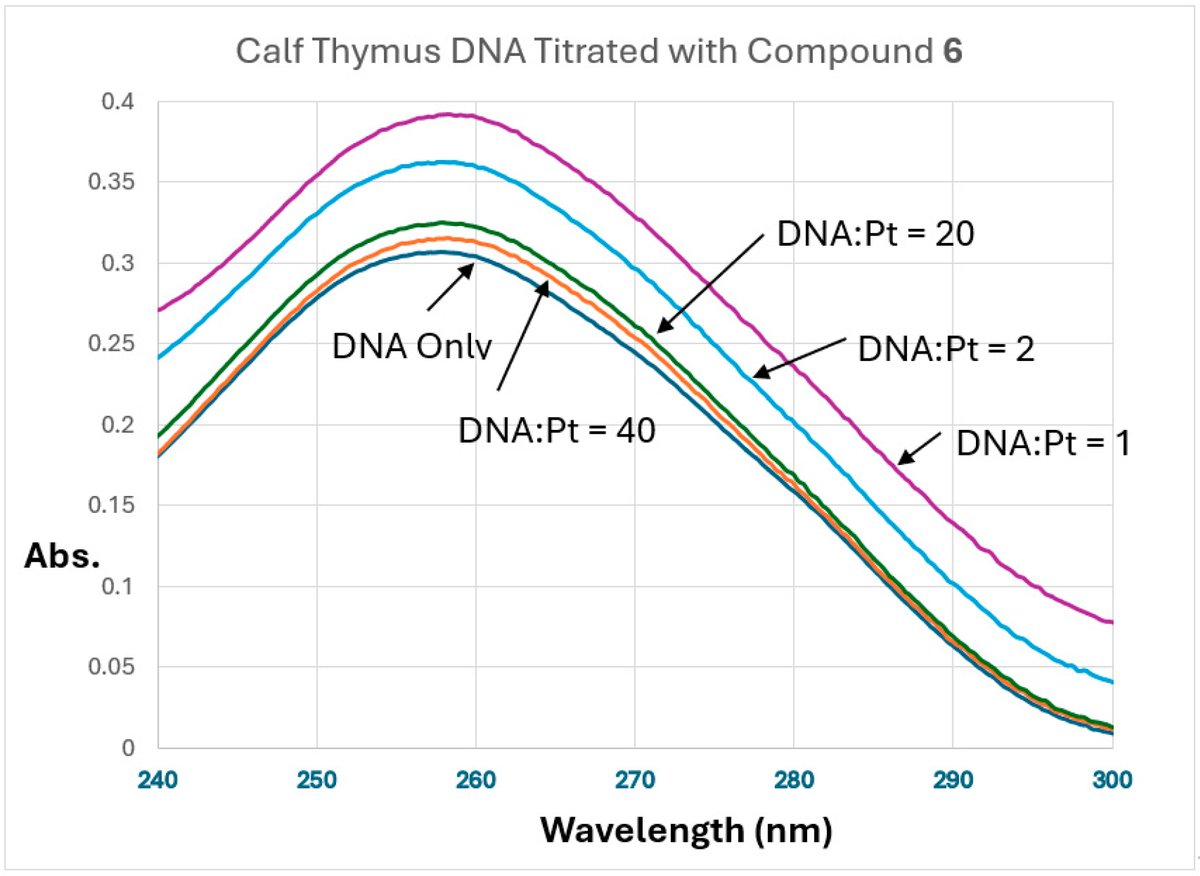
6
10
653

20 May 2025
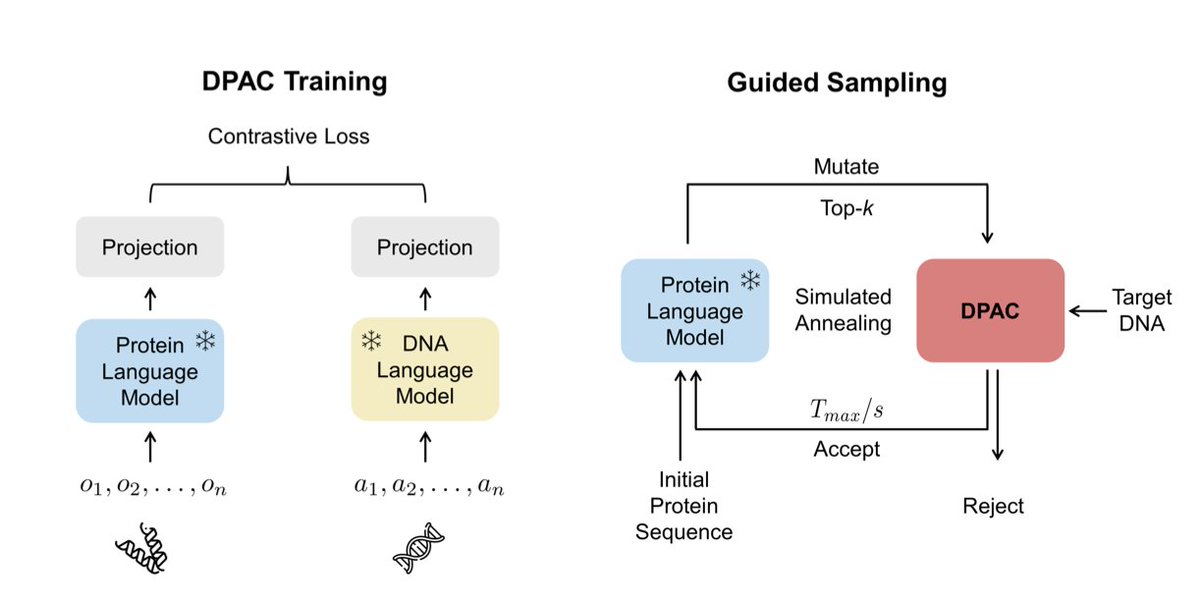
DPAC: Prediction and Design of Protein-DNA Interactions via Sequence-Based Contrastive Learning
1.This work introduces DPAC, a fully sequence-based framework that uses contrastive learning to predict and design protein-DNA interactions—bypassing the need for structural data and offering a scalable alternative to AlphaFold 3 and RosettaFoldNA.
2.DPAC aligns pre-trained protein (ESM2) and DNA (Nucleotide Transformer) language models into a shared latent space using a CLIP-style two-tower architecture, achieving high performance in both retrieval and design tasks.
3.On a low-identity benchmark (unseen protein-DNA pairs), DPAC significantly outperforms structure-based baselines (RFAA and RFNA), with an AUC of 0.617 vs. 0.546 and 0.456, respectively—demonstrating strong generalization.
4.The model supports inverse design using simulated annealing guided by DPAC logits, enabling the generation of protein sequences that maximize binding to target DNA motifs.
5.A case study on the Zif268 zinc finger shows that DPAC recovers up to 20% of binding affinity (ipTM score) in silico after introducing up to 50% sequence corruption, with restored amino acids spatially clustering near the DNA-binding interface.
6.DPAC-generated sequences also maintain biological plausibility, scoring favorably under ProGen2 perplexity metrics, and achieving comparable performance to LigandMPNN-based structure-guided design pipelines.
7.Extensive benchmarking confirms that DPAC’s performance is robust across embedding strategies, sequence identity levels, and temperature settings—reaching up to 0.85 AUC on high-identity sets with optimal encoder configurations.
8.DPAC also shows a statistically significant correlation between its logit scores and real binding affinity (Kd) across 68 protein-DNA binding datasets from PDBBind (p = 9e-5), supporting its use as a virtual screening tool.
9.Unlike structure-based models, DPAC is effective even for disordered or flexible DNA-binding proteins such as transcription factors, which pose challenges for co-folding approaches like AlphaFold 3.
10.By combining language model embeddings with contrastive learning and fast simulated annealing, DPAC sets a precedent for efficient, large-scale, and structure-free modeling of DNA-binding protein design.
11.The authors note future directions such as incorporating PTMs, exploring discrete diffusion-based generation, and applying DPAC to downstream applications in genome editing and transcriptional regulation.
💻Code: github.com/programmablebio/d…
📜Paper: biorxiv.org/content/10.1101/…
#ProteinDesign #DNABinding #ContrastiveLearning #LanguageModels #SyntheticBiology #AI4Science #GenomicEngineering #ComputationalBiology
3
4
581
12 May 2025
Assessment of nucleic acid structure prediction in CASP16
1.This paper presents a comprehensive assessment of RNA and DNA structure prediction in the CASP16 experiment, highlighting the stark contrast in predictive accuracy between nucleic acids and proteins, despite the rise of deep learning models.
2.Blind predictions were submitted for 42 targets by 65 groups from 46 institutions, covering a wide range of systems: RNA and DNA monomers, RNA-only multimers, nucleic acid-protein complexes, and NA-ligand complexes.
3.No prediction of a novel natural RNA structure reached a TM-score above 0.8, indicating that even with recent advances, atomic-resolution prediction of novel RNA structures remains out of reach.
4.Template-based modeling dominated the results: most accurate predictions required a closely related 3D template, and only a few targets, like OLE RNA, were accurately predicted without one—highlighting the dependency on structural homology.
5.Top-performing groups were all human expert teams (Vfold, GuangzhouRNA-human, and KiharaLab), outperforming automated servers like AlphaFold 3, especially for targets with shallow MSAs or no templates.
6.Secondary structure predictions were remarkably strong across the board, even outperforming traditional tools like ViennaRNA or EternaFold, suggesting reliable base-pair information is now extractable from sequence and MSA data alone.
7.However, predictions of pseudoknots, singlet base pairs, and tertiary interactions like A-minor motifs remained inconsistent or poor—critical gaps for enabling accurate 3D folding.
8.In ligand and complex structure predictions, performance was similarly limited unless prior structural templates existed. Complexes with novel binding interfaces were especially challenging.
9.One notable exception was the prediction of the OLE RNA structure, which had no close structural template but deep evolutionary information, suggesting future methods may leverage MSAs better for template-free modeling.
10.The paper introduces a rigorous multi-metric Z-score ranking system to evaluate groups across four categories: monomeric NA, RNA multimers, NA-protein, and NA-ligand complexes.
11.While server models like Yang-Server and AlphaFold 3 improved compared to previous CASPs, they still lagged behind human experts, especially in detecting complex or functionally important motifs.
12.Performance on tertiary motifs such as T-loops, UA-handles, and platforms was mixed, with automated methods performing comparably to humans in some cases, but failing on subtler structural features like intercalated bases.
13.The study advocates for better template identification, enhanced MSA construction, and refined evaluation metrics that go beyond global fold and include functional interaction motifs and quaternary contacts.
14.It also emphasizes the importance of future CASP assessments in benchmarking quaternary structure prediction, including symmetry and stoichiometry inference—areas where predictors still struggle.
15.Overall, CASP16 confirms that nucleic acid structure prediction is advancing, but still lags behind protein structure prediction in accuracy, automation, and reliability—particularly in the absence of experimental or homologous data.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #DNAbinding #CASP16 #StructuralBiology #ComputationalBiology #RNAfolding #NucleicAcids #DeepLearning #Bioinformatics #RNA3D #AlphaFold

1
668
12 May 2025
Assessment of nucleic acid structure prediction in CASP16
1.This paper presents a comprehensive assessment of RNA and DNA structure prediction in the CASP16 experiment, highlighting the stark contrast in predictive accuracy between nucleic acids and proteins, despite the rise of deep learning models.
2.Blind predictions were submitted for 42 targets by 65 groups from 46 institutions, covering a wide range of systems: RNA and DNA monomers, RNA-only multimers, nucleic acid-protein complexes, and NA-ligand complexes.
3.No prediction of a novel natural RNA structure reached a TM-score above 0.8, indicating that even with recent advances, atomic-resolution prediction of novel RNA structures remains out of reach.
4.Template-based modeling dominated the results: most accurate predictions required a closely related 3D template, and only a few targets, like OLE RNA, were accurately predicted without one—highlighting the dependency on structural homology.
5.Top-performing groups were all human expert teams (Vfold, GuangzhouRNA-human, and KiharaLab), outperforming automated servers like AlphaFold 3, especially for targets with shallow MSAs or no templates.
6.Secondary structure predictions were remarkably strong across the board, even outperforming traditional tools like ViennaRNA or EternaFold, suggesting reliable base-pair information is now extractable from sequence and MSA data alone.
7.However, predictions of pseudoknots, singlet base pairs, and tertiary interactions like A-minor motifs remained inconsistent or poor—critical gaps for enabling accurate 3D folding.
8.In ligand and complex structure predictions, performance was similarly limited unless prior structural templates existed. Complexes with novel binding interfaces were especially challenging.
9.One notable exception was the prediction of the OLE RNA structure, which had no close structural template but deep evolutionary information, suggesting future methods may leverage MSAs better for template-free modeling.
10.The paper introduces a rigorous multi-metric Z-score ranking system to evaluate groups across four categories: monomeric NA, RNA multimers, NA-protein, and NA-ligand complexes.
11.While server models like Yang-Server and AlphaFold 3 improved compared to previous CASPs, they still lagged behind human experts, especially in detecting complex or functionally important motifs.
12.Performance on tertiary motifs such as T-loops, UA-handles, and platforms was mixed, with automated methods performing comparably to humans in some cases, but failing on subtler structural features like intercalated bases.
13.The study advocates for better template identification, enhanced MSA construction, and refined evaluation metrics that go beyond global fold and include functional interaction motifs and quaternary contacts.
14.It also emphasizes the importance of future CASP assessments in benchmarking quaternary structure prediction, including symmetry and stoichiometry inference—areas where predictors still struggle.
15.Overall, CASP16 confirms that nucleic acid structure prediction is advancing, but still lags behind protein structure prediction in accuracy, automation, and reliability—particularly in the absence of experimental or homologous data.
📜Paper: biorxiv.org/content/10.1101/…
#RNAstructure #DNAbinding #CASP16 #StructuralBiology #ComputationalBiology #RNAfolding #NucleicAcids #DeepLearning #Bioinformatics #RNA3D #AlphaFold

1
2
6
1,032
8 May 2025
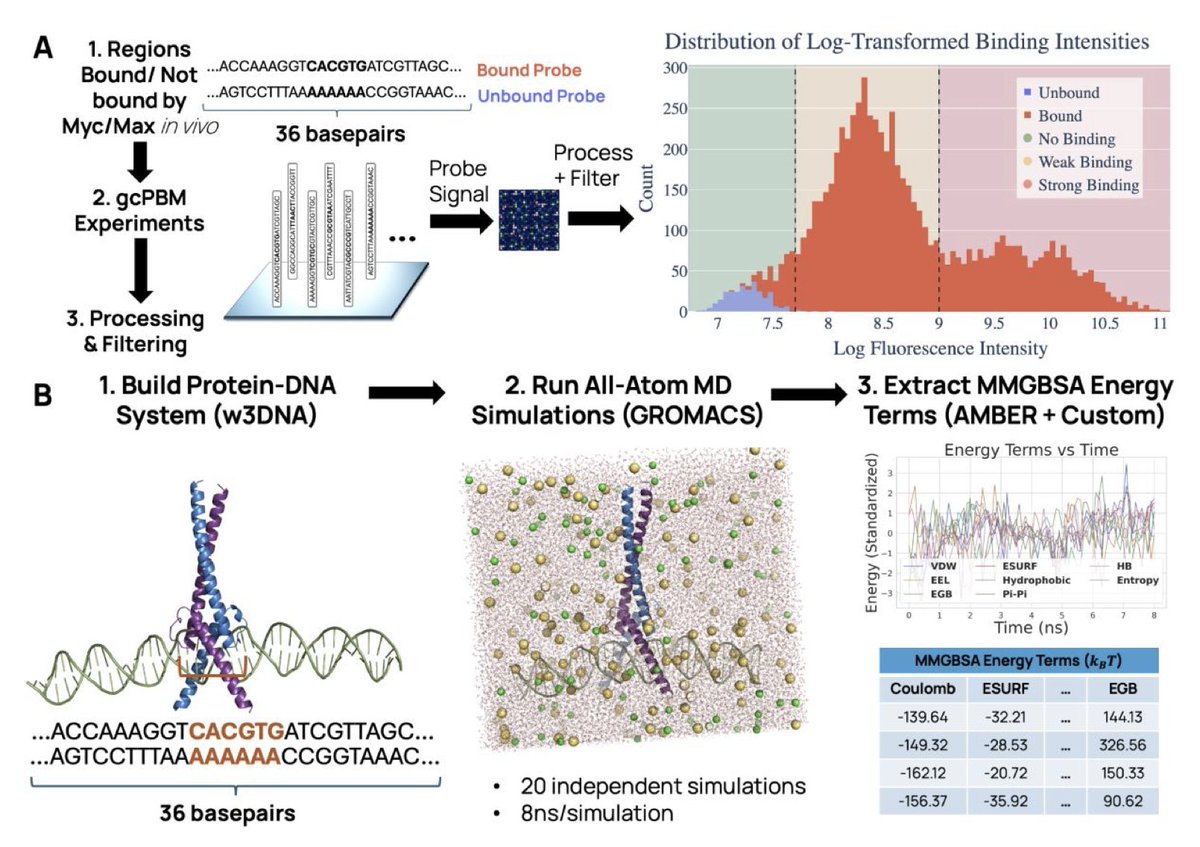
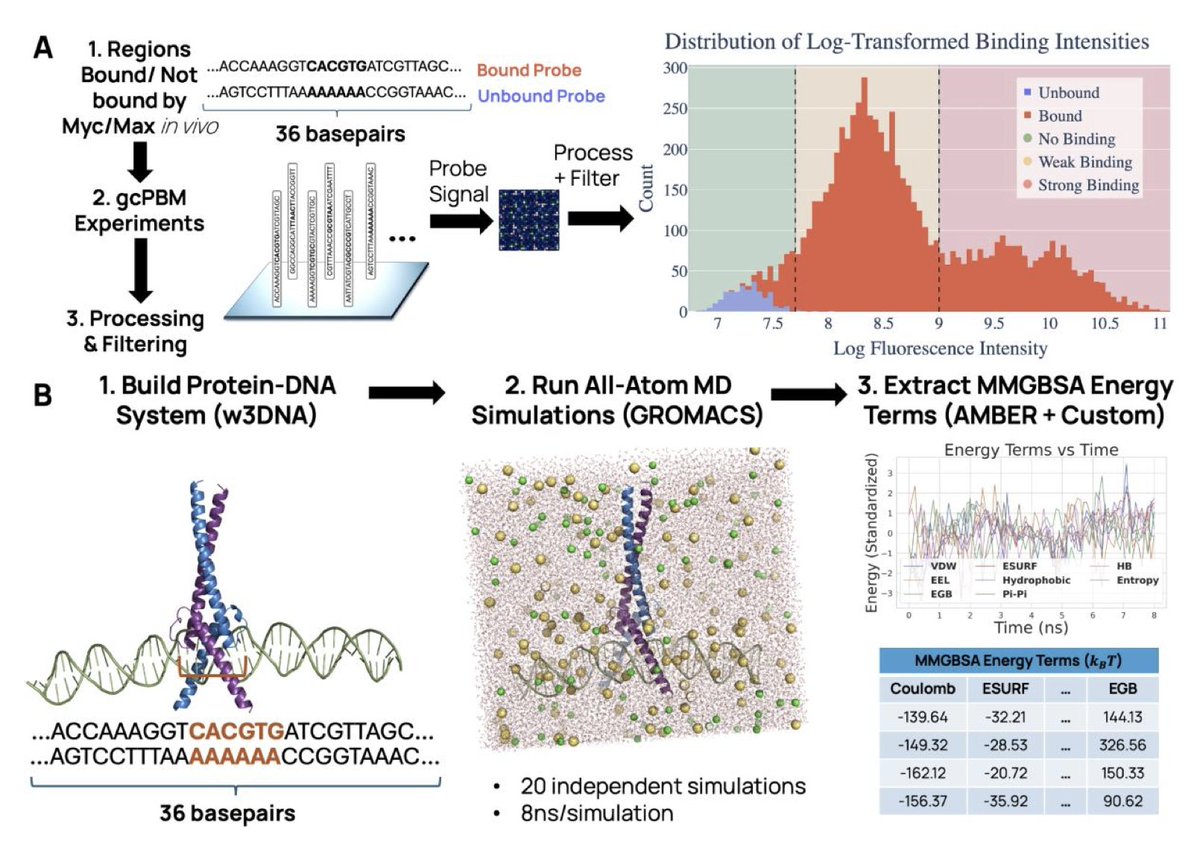
Combining Physics-Based Protein–DNA Energetics with Machine Learning to Predict Interpretable Transcription Factor-DNA Binding
1. This work presents a hybrid pipeline that integrates all-atom molecular dynamics simulations and MMGBSA energy decomposition with machine learning models to predict transcription factor (TF)–DNA binding affinities, achieving interpretability and strong predictive performance.
2. Focusing on the oncogenic Myc/Max dimer, the study constructs a dataset of 168 DNA sequences from genomic-context PBM assays and extracts 8 energy components per complex (e.g., van der Waals, electrostatics, hydrophobicity, hydrogen bonding) from ~3 million MD frames.
3. Standard MMGBSA linear summation gave only moderate correlation with experimental binding affinities (PCC ≈ 0.48), but integrating these features into ML models (RF, SVM, NN) significantly improved predictions (PCC ≈ 0.73, MSE reduced from 9.2 to <0.43).
4. Random Forest models revealed that hydrophobic interactions, van der Waals forces, and nonpolar solvation dominate the predictive signal—consistent with known shape complementarity principles in TF–DNA recognition.
5. Surprisingly, electrostatic and GB terms contributed minimally due to mutual cancellation, while hydrogen bonding effects initially appeared weak—until reparameterization with position-weight matrix (PWM) weights restored their importance and boosted accuracy.
6. Binary classification of binding vs non-binding sequences improved dramatically: F1 score rose from 0.26 with MMGBSA alone to 0.78 using ML-enhanced predictions, validating the added value of data-driven modeling over simple energy summation.
7. The study also tested linear regression and scrambled data controls. Linear models failed to generalize (PCC ≈ 0.12), and scrambled labels degraded performance to random, confirming that ML models capture real, sequence-specific biophysical features.
8. While the model performed well on core binding motifs (e.g., CACGTG E-box), it struggled to capture flanking sequence effects—suggesting opportunities for future improvement via dynamic contact modeling or sliding-based affinity estimators.
9. By integrating physics-informed descriptors and nonlinear learning, the framework achieves both mechanistic interpretability and predictive power—offering a promising route toward universal TF–DNA binding affinity prediction across sequence space.
10. The study highlights the synergy of simulation-based physical modeling and machine learning for understanding protein–nucleic acid interactions, with future extensions aimed at larger TF families and real-world genomic applications.
📜Paper: doi.org/10.26434/chemrxiv-20…
#TranscriptionFactors #DNABinding #MachineLearning #MMGBSA #MolecularDynamics #Bioinformatics #GeneRegulation #MycMax #PhysicsInformedML #TFDNA #ComputationalBiology
12
65
3,562
8 May 2025
Combining Physics-Based Protein–DNA Energetics with Machine Learning to Predict Interpretable Transcription Factor-DNA Binding
1. This work presents a hybrid pipeline that integrates all-atom molecular dynamics simulations and MMGBSA energy decomposition with machine learning models to predict transcription factor (TF)–DNA binding affinities, achieving interpretability and strong predictive performance.
2. Focusing on the oncogenic Myc/Max dimer, the study constructs a dataset of 168 DNA sequences from genomic-context PBM assays and extracts 8 energy components per complex (e.g., van der Waals, electrostatics, hydrophobicity, hydrogen bonding) from ~3 million MD frames.
3. Standard MMGBSA linear summation gave only moderate correlation with experimental binding affinities (PCC ≈ 0.48), but integrating these features into ML models (RF, SVM, NN) significantly improved predictions (PCC ≈ 0.73, MSE reduced from 9.2 to <0.43).
4. Random Forest models revealed that hydrophobic interactions, van der Waals forces, and nonpolar solvation dominate the predictive signal—consistent with known shape complementarity principles in TF–DNA recognition.
5. Surprisingly, electrostatic and GB terms contributed minimally due to mutual cancellation, while hydrogen bonding effects initially appeared weak—until reparameterization with position-weight matrix (PWM) weights restored their importance and boosted accuracy.
6. Binary classification of binding vs non-binding sequences improved dramatically: F1 score rose from 0.26 with MMGBSA alone to 0.78 using ML-enhanced predictions, validating the added value of data-driven modeling over simple energy summation.
7. The study also tested linear regression and scrambled data controls. Linear models failed to generalize (PCC ≈ 0.12), and scrambled labels degraded performance to random, confirming that ML models capture real, sequence-specific biophysical features.
8. While the model performed well on core binding motifs (e.g., CACGTG E-box), it struggled to capture flanking sequence effects—suggesting opportunities for future improvement via dynamic contact modeling or sliding-based affinity estimators.
9. By integrating physics-informed descriptors and nonlinear learning, the framework achieves both mechanistic interpretability and predictive power—offering a promising route toward universal TF–DNA binding affinity prediction across sequence space.
10. The study highlights the synergy of simulation-based physical modeling and machine learning for understanding protein–nucleic acid interactions, with future extensions aimed at larger TF families and real-world genomic applications.
📜Paper:
doi.org/10.26434/chemrxiv-20…
#TranscriptionFactors #DNABinding #MachineLearning #MMGBSA #MolecularDynamics #Bioinformatics #GeneRegulation #MycMax #PhysicsInformedML #TFDNA #ComputationalBiology
2
12
1,046
8 Apr 2025
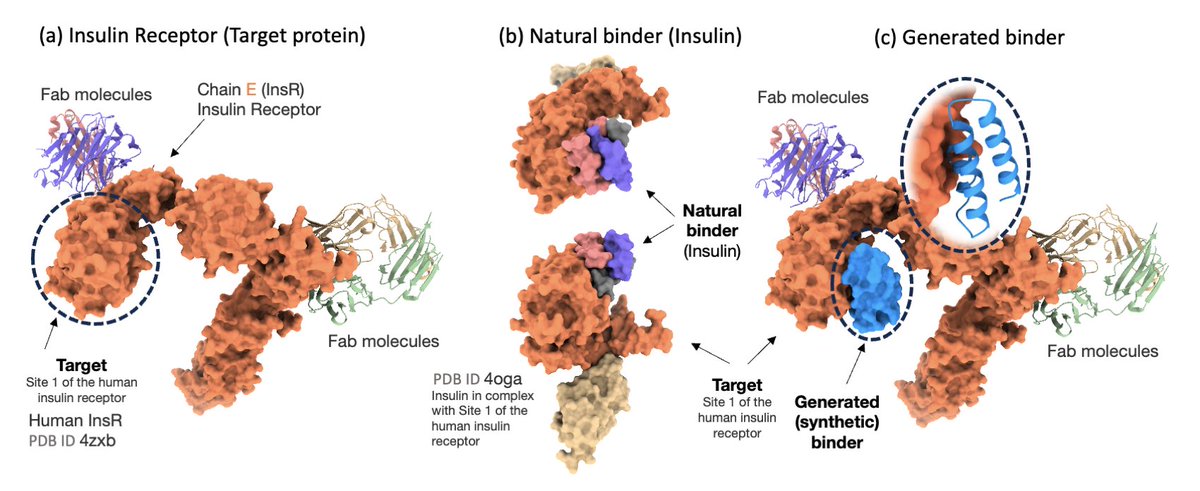
Prot42: a Novel Family of Protein Language Models for Target-aware Protein Binder Generation
1/ Prot42 introduces a powerful family of generative protein language models designed for sequence-based protein binder design, eliminating the need for 3D structural data or predefined binding sites. It opens new possibilities in therapeutic protein engineering and gene regulation.
2/ Unlike structure-based models like AlphaProteo or RFdiffusion, Prot42 uses an autoregressive, decoder-only transformer architecture to generate protein binders solely from sequence information. It supports sequences up to 8,192 amino acids, far beyond the 1k–2k limits of existing pLMs.
3/ The model achieves high binding specificity and affinity by learning conditional distributions over protein sequences, trained on pairs of target-binder interactions. It generates binders autoregressively, guided by stochastic sampling and affinity filtering.
4/ Prot42 excels not only in protein–protein binder generation but also in creating sequence-specific DNA-binding proteins. It integrates embeddings from Gene42 (genomic LMs) for context-aware generation, enabling applications in genome editing and synthetic biology.
5/ Experimental results show that Prot42-generated binders achieve competitive or superior predicted binding affinities compared to AlphaProteo, with lower dissociation constants (Kd) across multiple therapeutic targets including IL-7RA, PD-L1, TrkA, VEGF-A, and TNFα.
6/ The model was evaluated using the PEER benchmark suite across 14 tasks including protein function prediction, localization, structure, and interaction prediction. Prot42 consistently ranked among top performers, validating its broad generalization capabilities.
7/ Embeddings from Prot42 effectively capture subcellular localization and functional patterns, producing distinct clusters in t-SNE projections. This demonstrates its potential for use in downstream predictive tasks such as protein annotation, drug targeting, and synthetic design.
8/ For DNA-binding protein design, Prot42 uses a cross-attention mechanism to fuse DNA embeddings with protein representations, generating binders that interact with specific DNA motifs. This expands the model’s utility beyond protein-protein systems.
9/ Prot42 represents a generative leap in protein language modeling. Its scalable architecture, long-context support, and sequence-only operation redefine computational binder design, enabling rapid in silico exploration of functional protein space.
💻Code: huggingface.co/inceptionai
📜Paper: arxiv.org/abs/2504.04453
#ProteinDesign #Bioinformatics #DeepLearning #ProteinLanguageModel #AIinBiotech #DrugDiscovery #SyntheticBiology #DNABinding #ComputationalBiology #ProteinEngineering #FoundationModels #Transformer
1
9
49
3,996
8 Apr 2025
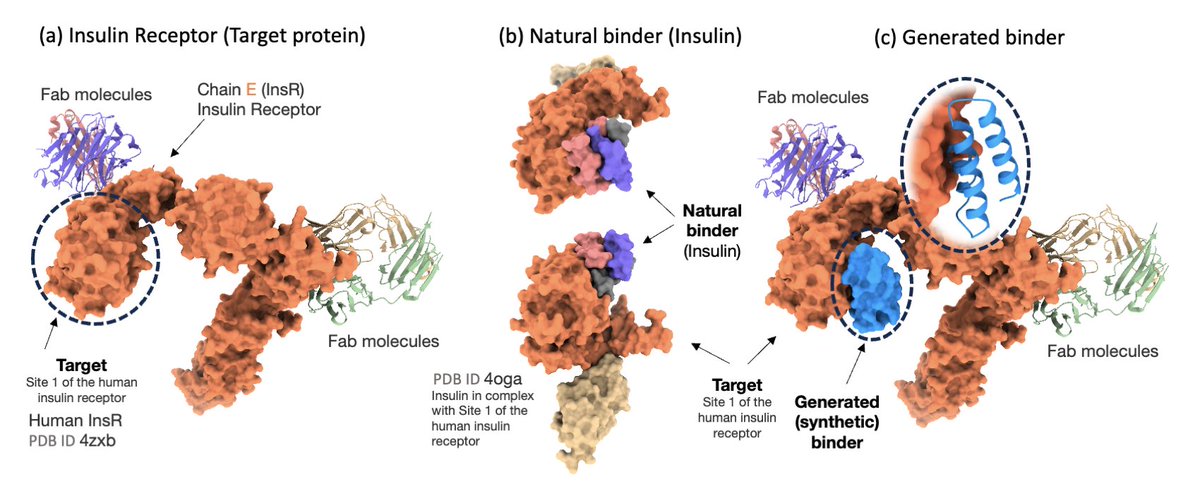
Prot42: a Novel Family of Protein Language Models for Target-aware Protein Binder Generation
1/ Prot42 introduces a powerful family of generative protein language models designed for sequence-based protein binder design, eliminating the need for 3D structural data or predefined binding sites. It opens new possibilities in therapeutic protein engineering and gene regulation.
2/ Unlike structure-based models like AlphaProteo or RFdiffusion, Prot42 uses an autoregressive, decoder-only transformer architecture to generate protein binders solely from sequence information. It supports sequences up to 8,192 amino acids, far beyond the 1k–2k limits of existing pLMs.
3/ The model achieves high binding specificity and affinity by learning conditional distributions over protein sequences, trained on pairs of target-binder interactions. It generates binders autoregressively, guided by stochastic sampling and affinity filtering.
4/ Prot42 excels not only in protein–protein binder generation but also in creating sequence-specific DNA-binding proteins. It integrates embeddings from Gene42 (genomic LMs) for context-aware generation, enabling applications in genome editing and synthetic biology.
5/ Experimental results show that Prot42-generated binders achieve competitive or superior predicted binding affinities compared to AlphaProteo, with lower dissociation constants (Kd) across multiple therapeutic targets including IL-7RA, PD-L1, TrkA, VEGF-A, and TNFα.
6/ The model was evaluated using the PEER benchmark suite across 14 tasks including protein function prediction, localization, structure, and interaction prediction. Prot42 consistently ranked among top performers, validating its broad generalization capabilities.
7/ Embeddings from Prot42 effectively capture subcellular localization and functional patterns, producing distinct clusters in t-SNE projections. This demonstrates its potential for use in downstream predictive tasks such as protein annotation, drug targeting, and synthetic design.
8/ For DNA-binding protein design, Prot42 uses a cross-attention mechanism to fuse DNA embeddings with protein representations, generating binders that interact with specific DNA motifs. This expands the model’s utility beyond protein-protein systems.
9/ Prot42 represents a generative leap in protein language modeling. Its scalable architecture, long-context support, and sequence-only operation redefine computational binder design, enabling rapid in silico exploration of functional protein space.
💻Code: huggingface.co/inceptionai
📜Paper: arxiv.org/abs/2504.04453
#ProteinDesign #Bioinformatics #DeepLearning #ProteinLanguageModel #AIinBiotech #DrugDiscovery #SyntheticBiology #DNABinding #ComputationalBiology #ProteinEngineering #FoundationModels #Transformer
1
24
98
5,664
6 Apr 2025
TransBind: A Multi-Objective Molecular Generation Method Based on Pareto Algorithm and Monte Carlo @NatureComms
1/ TransBind introduces an innovative approach to predicting DNA-binding proteins and residues, using a combination of transformer-based protein language models and deep learning. This method eliminates the need for MSA-based evolutionary features, making it computationally efficient.
2/ Unlike traditional methods that rely on PSSMs or HMMs, TransBind utilizes ProtTrans, a pretrained protein language model, to generate highly accurate features directly from protein sequences without requiring multiple sequence alignments (MSAs).
3/ The model achieves high precision in both DNA-binding protein and residue prediction tasks. TransBind significantly improves upon previous methods in sensitivity, specificity, and MCC (Matthews Correlation Coefficient), offering a balanced approach to DNA-binding prediction.
4/ A standout feature of TransBind is its handling of class imbalance in training data. By using a weighted loss function, it enhances model sensitivity without compromising accuracy, even on datasets with a disproportionate number of non-binding residues.
5/ TransBind demonstrates its advantage by outperforming existing state-of-the-art methods in terms of computational speed and accuracy across multiple benchmark datasets, including PDNA and RNA datasets, proving its practical utility in protein sequence-based predictions.
6/ One of the key innovations is the ability to predict DNA-binding residues without the need for structural information, making TransBind particularly useful for orphan proteins or those with sparse homologous data, such as antibodies.
7/ With an emphasis on real-world applications, TransBind is available as an open-source tool and web server, making it accessible for researchers to use and integrate into their DNA-binding protein research workflows.
📜Paper: nature.com/articles/s42003-0…
#ComputationalBiology #ProteinPrediction #DeepLearning #MachineLearning #Bioinformatics #DNABinding

1
8
1,016

11 Mar 2025
Specificity landscapes of 40 R2R3‐MYBs reveal how paralogs target different cis‐elements by homodimeric binding onlinelibrary.wiley.com/doi/… #DNAbinding @WileyBiomedical
8
7
1,894

7 Sep 2024
nature.com/articles/s41467-0…
Our paper is out in Nat. Commun.! 🎉 An amazing collab between the Aggarwal, Fang, Filizola, and Shapiro labs. We show a mechanism of #DNAbinding wherein each monomer of M.BceJIV #methyltransferase helps to recognize two #DNAs!
#epigenetics #methylation

4
4
17
3,944

15 Jul 2024
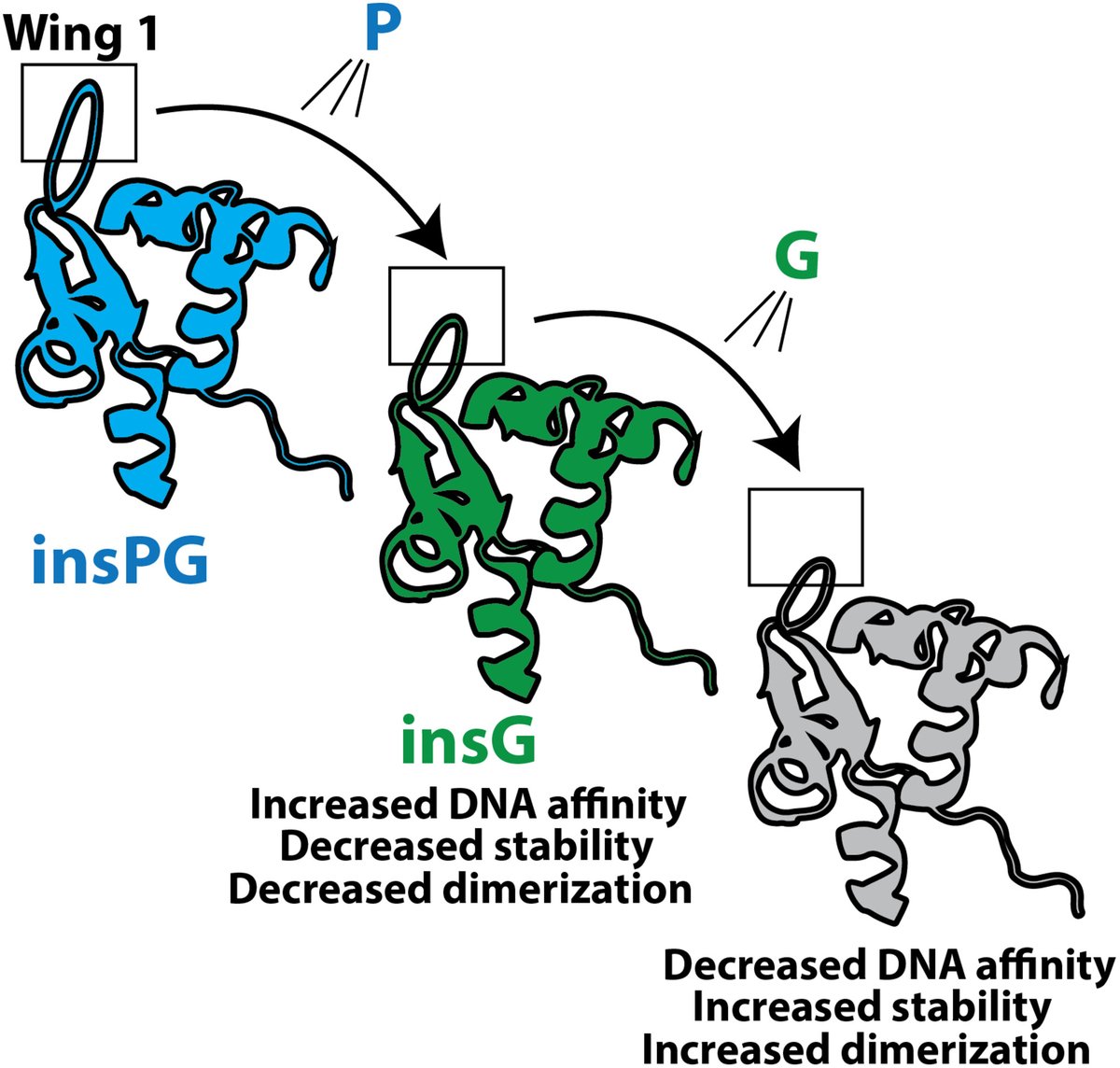
Lost in evolution
🤔Why did the human FoxP1 #transcriptionfactor lose a Pro–Gly sequence in its #DNAbinding domain during evolution?
👉Find out in this study by Exequiel Medina @uchile & team
➡bit.ly/4eYoAMA
#proteinevolution #proteinproteininteractions
2
2
333

30 Oct 2023
2
254

19 Jul 2023
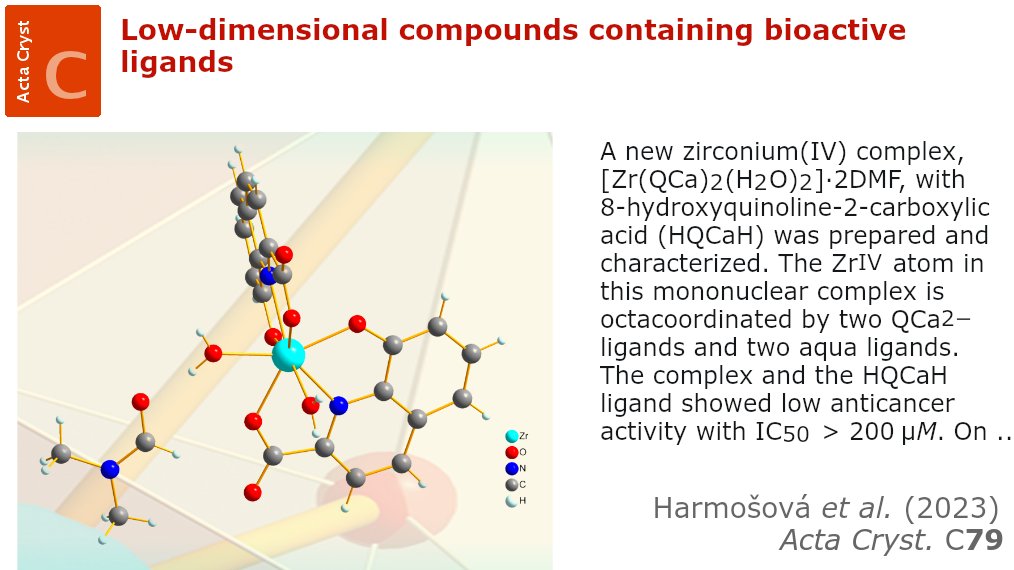
Low-dimensional compounds containing bioactive ligands @ActaCrystC @IUCr doi.org/10.1107/S20532296230… @UPJS_kosice @CzechAcademy @FZU_AVCR #VEGA #APVV @EU_Regional @msmtcr #cytotoxicity #zinc #quinoline #carboxylicacid #bioactive #DNAbinding #HSAbinding #crystallography
1
5
212

Zinc fingers tune chromatin ... like potters' fingers craft clay!
Please consider our Special Issue of Cancers "Zinc-Finger Proteins in Cancer" IF=6.6 @MDPIOpenAccess @CTCF_Papers @DNA_RNA_Uni @DNAbinding @DNAmPapers @boris_ctcf @TF_binding_bot mdpi.com/journal/cancers/spe…

1
6

5 Jan 2022
Mess or science? This is how we do in (bio)inorganic chemistry! #PhDLife #inorganicchem #dnabinding #dnainteraction

3

24 Nov 2021
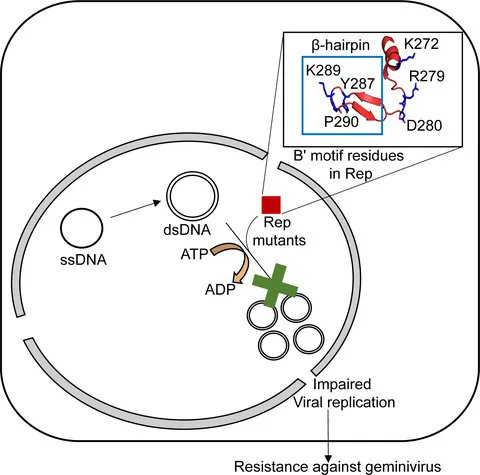
Functional implications of residues of the B′ motif of geminivirus replication initiator protein in its helicase activity
buff.ly/3FzTw39
By Supriya Chakraborty and colleagues @JNU_official_50
#Geminivirus #ssDNAvirus #viralreplication #Helicase #ATPhydrolysis #DNAbinding
2

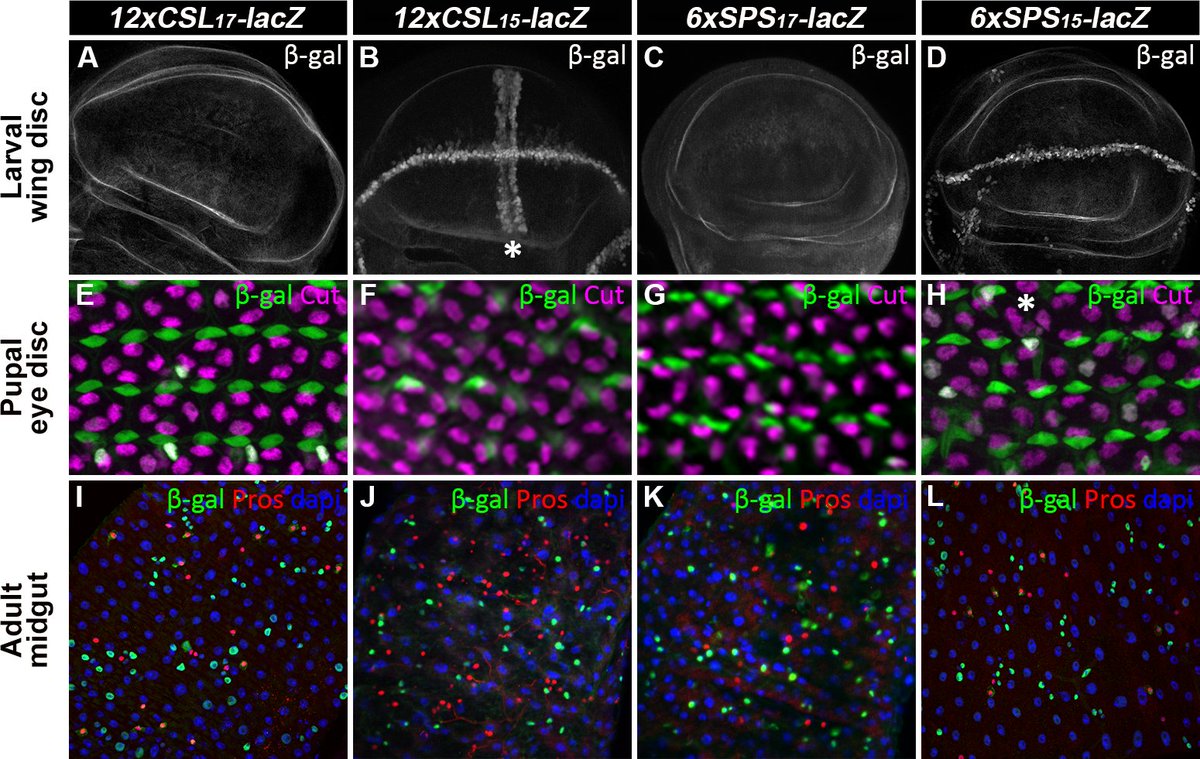
Enhancers with cooperative Notch binding sites are more resistant to regulation by the Hairless co-repressor
New in @PLOSGenetics w/Matt Weirauch @CincyChildrens @UCHealthNews: journals.plos.org/plosgeneti…
#DNAbinding #CellSignaling #GeneExpression
1
3
27 May 2021
🌟 Editor's Choice 🌟
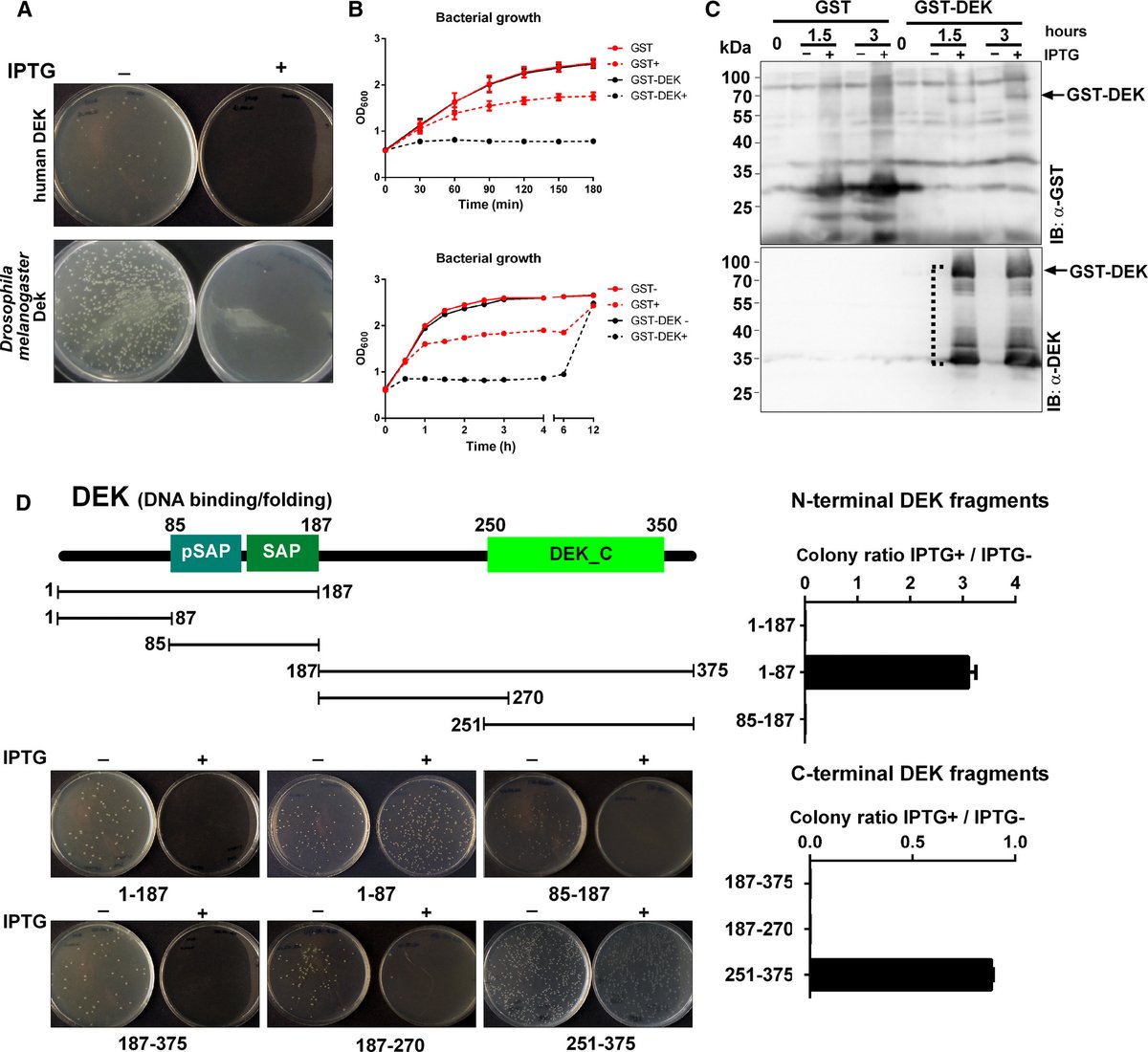
Bacterial Growth Inhibition Screen (BGIS): a new method for studying #proteinfunction by harnessing #recombinantprotein toxicity in #Ecoli
🔍 By Ferdinand Kappes & team
➡bit.ly/3yLFfxQ
#proteinanalysis #oncoprotein #DNAbinding #RNAbinding
1
5

28 Apr 2021
Don't miss our talk starting at 1:30pm today at #ExpBio to discover how dynamic single-molecule technologies can help you obtain direct proof of the mechanisms of #DNABinding proteins and observe the stepwise assembly of the #biological complex.
➡️bit.ly/3gKXQmV
#EB2021
27 Apr 2021

Getting direct proof of molecular mechanisms of #DNABindingProteins is not impossible anymore! Wanna learn how? We show you in our talk at the upcoming #ExpBio/#EB2021. Register now➡️ bit.ly/3wWuLdS
2
3