Multilabel prediction of virus target proteins via multimodal graph representation learning
1 MultiVTP reframes virus target protein (VTP) identification as a multilabel host-protein problem: a single human protein can be targeted by multiple viruses, and prediction can be done using only intrinsic host information (no viral proteins required).
2 The core idea is to learn host susceptibility signals from the human PPI network plus multimodal protein descriptors, then output a vector of virus-specific targeting probabilities per host protein (species-level and family-level labels).
3 Architecture overview: (i) multi-view subgraph sampling around each query protein via repeated random walks, (ii) feature extraction (network topology multimodal), (iii) Graphormer-based integration inside each subgraph, (iv) Progressive Layered Extraction (PLE) to separate shared vs virus-specific binding patterns for multilabel prediction.
4 Network topology is treated at two scales: global roles via node2vec embeddings (256D) and local positions via shortest-path distance encodings used as attention bias in Graphormer; ablations show global topology and the Graphormer module are the largest performance drivers.
5 Multimodal protein features combine (a) traditional curated features (sequence composition, evolutionary conservation metrics like dN/dS and protein age, predicted secondary structure/solvent accessibility, and classic network centralities), (b) sequence embeddings from ESM2, and (c) functional embeddings from GO text encoded by PubMedBERT and aggregated with a GCN over a GO-similarity graph.
6 The PLE multilabel head explicitly models cross-virus commonalities (shared expert) and virus-specific signatures (task experts gating), improving over simpler multilabel strategies (binary relevance / classifier chains / label powerset) and over replacing PLE with a plain MLP.
7 Interpretability: Graphormer self-attention assigns higher attention to VTPs than non-VTPs; proteins with high attention are enriched for host–virus interaction, innate immunity, and antiviral defense processes, suggesting the model prioritizes biologically relevant neighborhoods rather than arbitrary graph proximity.
8 Benchmarking highlights: MultiVTP outperforms host-only HIVPRE for HIV-1 target prediction (reported gains in both AUC and AUPR), beats multiple multilabel baselines (MLP, XGB, RF, SVM with standard multilabel strategies), and remains comparatively robust when training positives are downsampled.
9 Few-shot setting: for viruses with only 20–100 known targets, training-from-scratch already outperforms strong baselines, while fine-tuning a pre-trained MultiVTP model yields large gains (example noted: AAV2 AUPR improvement from scratch to fine-tuned), supporting adaptation to emerging/understudied viruses.
10 Human proteome application: scoring 20,270 UniProt proteins enables systematic nomination of novel VTP candidates per virus and candidates targeted by multiple viruses (MVTPs). Case studies (e.g., H1N1, HIV-1) show predicted candidates tend to connect to known VTPs in the PPI network and enrich known and additional pathways; MVTP candidates show higher conservation and central network positions, suggesting potential as broad-spectrum antiviral targets.
💻Code: github.com/hzau-liulab/Multi…
📜Paper: doi.org/10.1371/journal.pcbi…
#ComputationalBiology #Bioinformatics #GraphNeuralNetworks #GraphTransformer #MultiLabelLearning #HostVirusInteractions #Proteomics #SystemsBiology #MachineLearning #DeepLearning
3
9
1,095
A Comprehensive Atlas and Machine-Learning Framework for Predicting IDR-Protein Binding Affinity
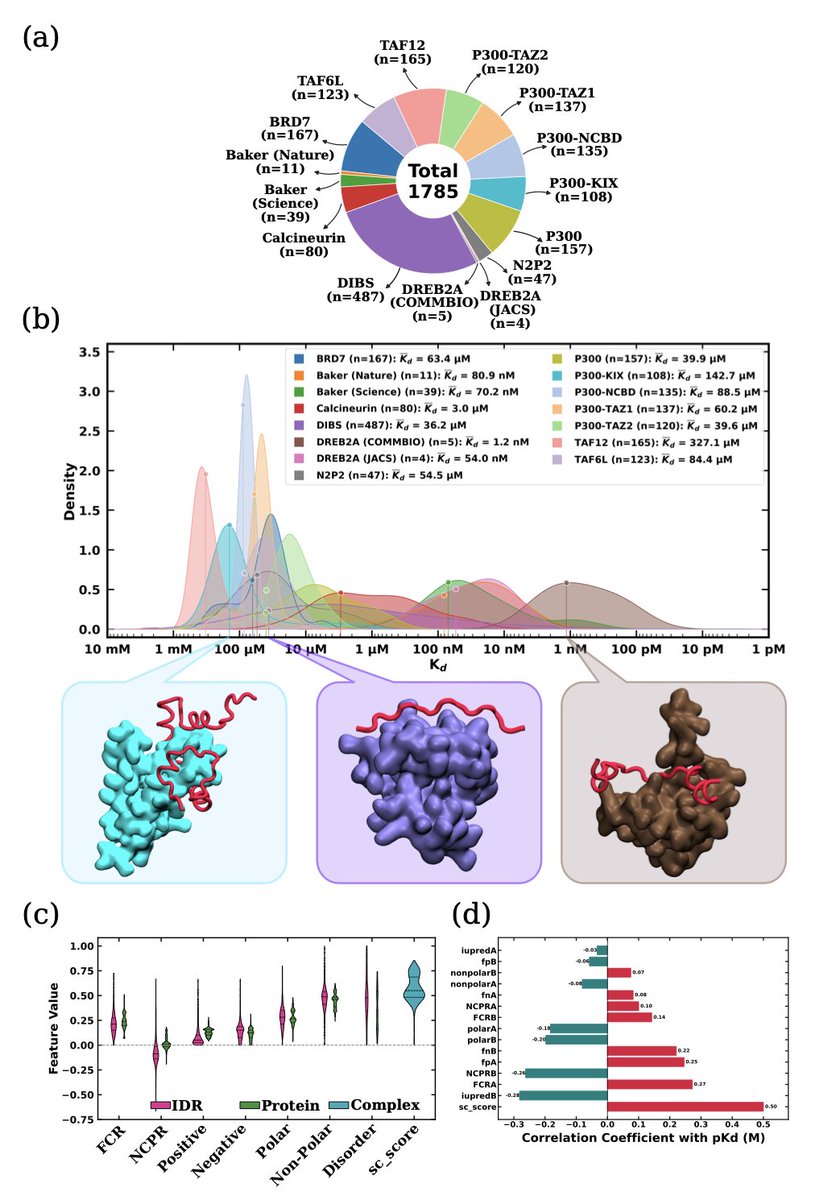
1. The study introduces IBPC-Kd, a curated dataset of 1,785 IDR-ordered protein complexes with experimentally measured dissociation constants (Kd), spanning more than six orders of magnitude in affinity.
2. The authors developed IDRBindNet, a graph-transformer model that integrates protein language model embeddings with interface geometry and residue-level chemical context to predict binding affinities from IDR-partner complexes.
3. IDRBindNet achieves state-of-the-art accuracy with R² up to 0.911 on held-out complexes, significantly outperforming existing generic protein-protein affinity predictors like PPAP and ProAffinity-GNN.
4. The model's attention mechanism implicitly learns biologically relevant patterns corresponding to partner disorder and geometric fit, without explicit feature engineering.
5. Key biophysical insights reveal that global interfacial shape complementarity is the strongest correlate of binding affinity, followed by structural order of the partner surface and systematic electrostatic asymmetry between IDRs and their binders.
6. The framework successfully generalizes to external validation on de novo designed binders targeting the human surfaceome, showing robust performance across nanomolar-to-micromolar affinities even on out-of-distribution data.
7. Unsupervised clustering of the dataset reveals distinct interaction regimes characterized by different combinations of physicochemical properties, with shape complementarity emerging as the dominant determinant at the cluster level (r = 0.90).
8. Residue-level analysis shows that burial of small, polar residues—particularly serine and glycine—exhibits the strongest correlations with shape complementarity, providing steric flexibility for optimal interfacial packing.
9. The study addresses a critical gap in quantitative affinity data for disordered protein complexes, as existing databases like DIBS contained only 487 entries with Kd values, insufficient for training robust machine learning models.
10. The authors provide an easy-to-run implementation on GitHub, enabling researchers to obtain predicted Kd values by simply supplying sequences and structures of binding partners.
💻Code: github.com/JMLab-tifrh/IDRBi…
📜Paper: biorxiv.org/content/10.64898…
#IntrinsicallyDisorderedProteins #ProteinBinding #MachineLearning #GraphTransformer #AlphaFold #ProteinDesign #Bioinformatics #ComputationalBiology #ProteinProteinInteractions #DrugDiscovery
4
16
1,475
Amortized Molecular Optimization via Group Relative Policy Optimization
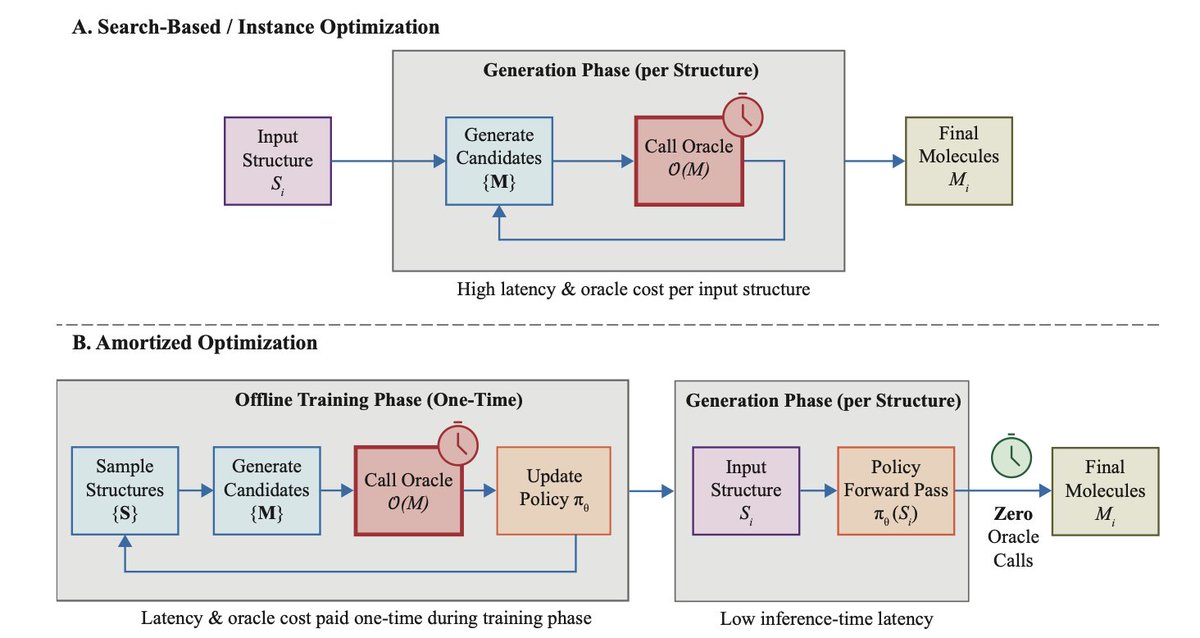
1. The paper introduces GRXForm, a novel amortized optimization framework that eliminates the need for expensive iterative oracle calls during inference, enabling single-pass molecular optimization for high-throughput drug discovery.
2. The key innovation is adapting Group Relative Policy Optimization (GRPO) from large language model training to molecular design, which addresses the critical challenge of high variance caused by heterogeneous scaffold difficulty.
3. Unlike instance optimizers like genetic algorithms and discrete diffusion models that require thousands of oracle evaluations per molecule, GRXForm front-loads computation into training, achieving zero oracle calls at inference time.
4. GRPO works by sampling groups of trajectories from the same starting scaffold and normalizing rewards relative to the group mean, creating dynamic baselines that stabilize learning across both easy and hard optimization tasks.
5. The method builds on a Graph Transformer architecture with hierarchical action space and valence-based masking to ensure chemical validity, pretrained on ChEMBL to capture chemical plausibility before reinforcement learning fine-tuning.
6. On the kinase scaffold decoration benchmark with strict multi-parameter optimization criteria, GRXForm achieved 17.8% success rate while all baselines including Mol GA and GenMol failed to produce a single valid molecule.
7. The ablation study confirms that GRPO significantly outperforms standard REINFORCE with global baseline (9.1% vs 17.8% success rate), with empirical evidence showing dramatically reduced advantage signal variance during training.
8. In a few-shot prodrug design case study, GRXForm successfully transferred lipidization rules to unseen complex drugs like Naltrexone where the REINFORCE baseline completely failed.
9. On the PMO benchmark, GRXForm achieved second-place overall performance with 16.433 cumulative AUC, demonstrating competitive optimization power despite being designed for amortized inference rather than instance search.
10. The amortization break-even analysis shows GRXForm becomes more efficient than state-of-the-art instance optimizers after processing just 5 to 250 scaffolds, with inference latency reduced from minutes to milliseconds per molecule.
💻Code: github.com/Hash-hh/GRXForm
📜Paper: arxiv.org/abs/2602.12162
#MolecularOptimization #DrugDiscovery #ReinforcementLearning #GraphTransformer #Cheminformatics #AIforScience #ComputationalChemistry #GenerativeAI #AmortizedInference #GRPO
2
5
958

17 Dec 2025
GlycanGT: A Foundation Model for Glycan Graphs with Pretrained Representation and Generative Learning
1. GlycanGT is a novel foundation model for glycans, leveraging a graph transformer architecture to address the complexity and diversity of glycan structures. This model outperforms existing methods in multiple benchmark tasks, demonstrating superior performance in capturing both local and global structural patterns of glycans.
2. The model employs masked language modeling (MLM) for pretraining, enabling GlycanGT to predict masked monosaccharides and glycosidic bonds. This innovative approach allows for the accurate reconstruction of ambiguous or incomplete glycan sequences, with over 80% top-5 accuracy in predictions.
3. GlycanGT's embeddings form biologically meaningful clusters, recovering known N- and O-glycan categories. This capability not only enhances the understanding of glycan functions but also provides a powerful tool for glycan database expansion and translational applications.
4. The study highlights GlycanGT's ability to capture global structural patterns, overcoming limitations of conventional graph neural networks such as over-smoothing and over-squashing. This advancement is crucial for tasks like taxonomy classification and immunogenicity prediction, where capturing long-range dependencies is essential.
5. The pretraining dataset includes over 80,000 glycans from GlyCosmos, ensuring a robust and diverse training environment. GlycanGT's performance is expected to improve further with larger and more standardized datasets, paving the way for future advancements in glycan research.
6. The pretrained GlycanGT model weights and usage scripts are publicly available on Hugging Face, making it accessible for researchers to fine-tune or adapt to additional glycan-related datasets and downstream tasks. This open-source approach fosters collaboration and accelerates the development of glycan-based applications.
💻Code: huggingface.co/Akikitani295/…
📜Paper: biorxiv.org/content/10.64898…
#GlycanResearch #GraphTransformer #Bioinformatics #PretrainedModels #GlycanGT #ComputationalBiology

2
7
1,197
30 May 2025
PSBench: a large-scale benchmark for estimating the accuracy of protein complex structural models
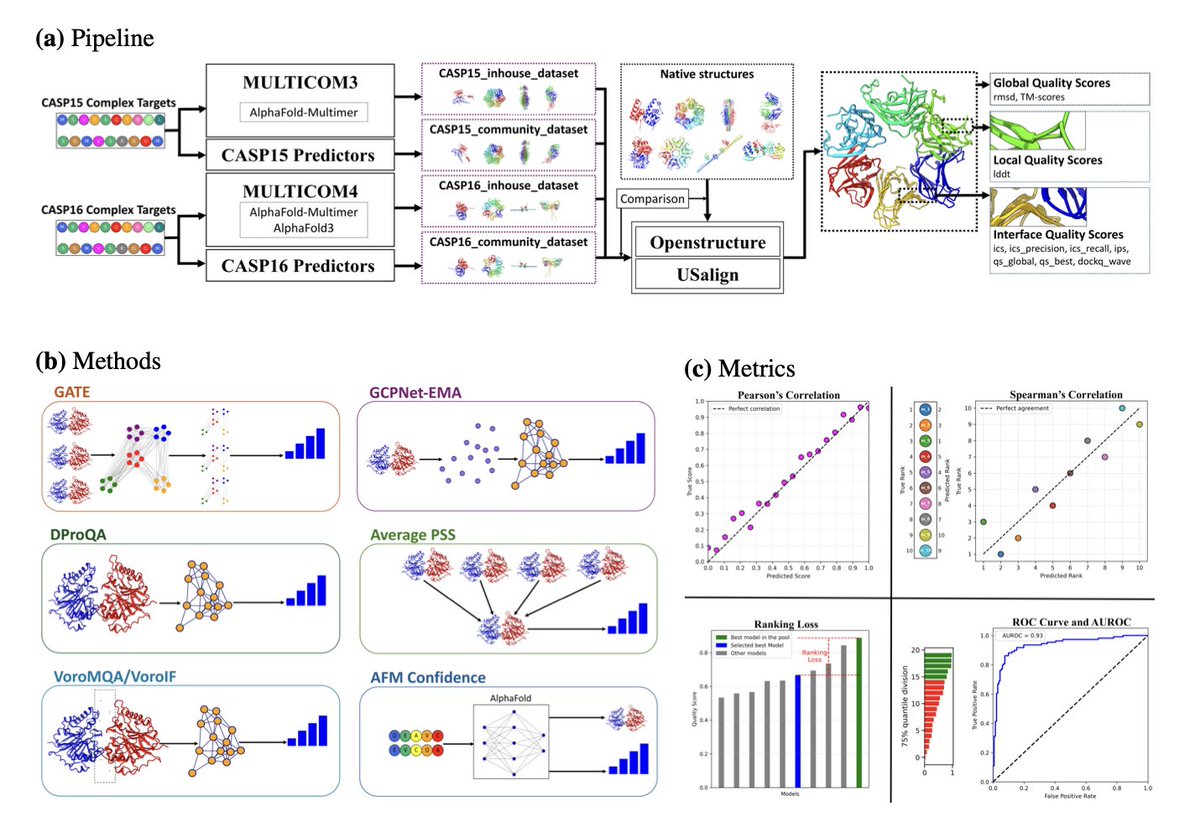
1.Accurately estimating the quality of predicted protein complex structures remains a major challenge, even with powerful tools like AlphaFold. PSBench addresses this by providing a large, diverse, and labeled benchmark dataset for evaluating Estimation of Model Accuracy (EMA) methods.
2.PSBench includes over one million predicted structural models from CASP15 and CASP16, generated in blind prediction settings. Each model is annotated with 10 distinct quality scores, covering global, local, and interface accuracy—offering the most comprehensive set of labels for EMA to date.
3.Existing EMA datasets are small, outdated, and often limited to simple complexes or docking-generated models. PSBench uniquely focuses on models produced by modern predictors like AlphaFold2-Multimer and AlphaFold3, making it highly relevant for today's structure prediction landscape.
4.To demonstrate PSBench’s practical utility, the authors developed GATE—a graph transformer-based EMA model. GATE was blindly evaluated in CASP16 and ranked among the top EMA predictors, proving the benchmark’s effectiveness for training state-of-the-art methods.
5.PSBench is organized into four datasets: CASP15_inhouse, CASP15_community, CASP16_inhouse, and CASP16_community. Together they cover 79 diverse protein complexes with varied stoichiometries, lengths (96–8460 residues), and modeling challenges.
6.The benchmark includes a complete evaluation framework with four metrics—Pearson and Spearman correlations, ranking loss, and AUROC—plus six baseline EMA methods for rigorous comparison. All evaluation tools are open-source and packaged with the benchmark.
7.The CASP16 results show GATE significantly outperforms other EMA methods in selecting high-quality AlphaFold-generated models, both in terms of ranking accuracy and classification performance. This success helped MULTICOM4, which used GATE, place among the top predictors in CASP16.
8.Unlike older datasets, PSBench provides a real-world setting: structural models are predicted before native structures are available, emulating realistic prediction scenarios and avoiding data leakage during training.
9.Each model in PSBench is stored in PDB format and includes additional features (e.g., AlphaFold confidence scores, inter-chain errors), enabling the development of more sophisticated EMA models using a richer feature set.
10.The benchmark is expandable—researchers can use the included annotation pipeline to label new models and contribute to PSBench, aiming to build a community-driven resource similar to ImageNet for structural biology.
💻Code: github.com/BioinfoMachineLea… 📜Paper: arxiv.org/abs/2505.22674v1 #ProteinStructure #AlphaFold #GraphTransformer #MachineLearning #StructuralBiology #CASP16 #Bioinformatics #ProteinComplexes #EMA #Benchmarking
3
19
1,298
30 May 2025
PSBench: a large-scale benchmark for estimating the accuracy of protein complex structural models
1.Accurately estimating the quality of predicted protein complex structures remains a major challenge, even with powerful tools like AlphaFold. PSBench addresses this by providing a large, diverse, and labeled benchmark dataset for evaluating Estimation of Model Accuracy (EMA) methods.
2.PSBench includes over one million predicted structural models from CASP15 and CASP16, generated in blind prediction settings. Each model is annotated with 10 distinct quality scores, covering global, local, and interface accuracy—offering the most comprehensive set of labels for EMA to date.
3.Existing EMA datasets are small, outdated, and often limited to simple complexes or docking-generated models. PSBench uniquely focuses on models produced by modern predictors like AlphaFold2-Multimer and AlphaFold3, making it highly relevant for today's structure prediction landscape.
4.To demonstrate PSBench’s practical utility, the authors developed GATE—a graph transformer-based EMA model. GATE was blindly evaluated in CASP16 and ranked among the top EMA predictors, proving the benchmark’s effectiveness for training state-of-the-art methods.
5.PSBench is organized into four datasets: CASP15_inhouse, CASP15_community, CASP16_inhouse, and CASP16_community. Together they cover 79 diverse protein complexes with varied stoichiometries, lengths (96–8460 residues), and modeling challenges.
6.The benchmark includes a complete evaluation framework with four metrics—Pearson and Spearman correlations, ranking loss, and AUROC—plus six baseline EMA methods for rigorous comparison. All evaluation tools are open-source and packaged with the benchmark.
7.The CASP16 results show GATE significantly outperforms other EMA methods in selecting high-quality AlphaFold-generated models, both in terms of ranking accuracy and classification performance. This success helped MULTICOM4, which used GATE, place among the top predictors in CASP16.
8.Unlike older datasets, PSBench provides a real-world setting: structural models are predicted before native structures are available, emulating realistic prediction scenarios and avoiding data leakage during training.
9.Each model in PSBench is stored in PDB format and includes additional features (e.g., AlphaFold confidence scores, inter-chain errors), enabling the development of more sophisticated EMA models using a richer feature set.
10.The benchmark is expandable—researchers can use the included annotation pipeline to label new models and contribute to PSBench, aiming to build a community-driven resource similar to ImageNet for structural biology.
💻Code: github.com/BioinfoMachineLea…
📜Paper: arxiv.org/abs/2505.22674v1
#ProteinStructure #AlphaFold #GraphTransformer #MachineLearning #StructuralBiology #CASP16 #Bioinformatics #ProteinComplexes #EMA #Benchmarking

1
4
546

28 May 2025
Predicting lymphatic transport potential using graph transformer based on limited historical data from in vivo studies.
| Sifei Han, China Pharmaceutical Univ. |
[50 days' free access]
#LymphaticTransport #ArtificialIntelligence #GraphTransformer
kwnsfk27.r.eu-west-1.awstrac…

2
216
11 May 2025
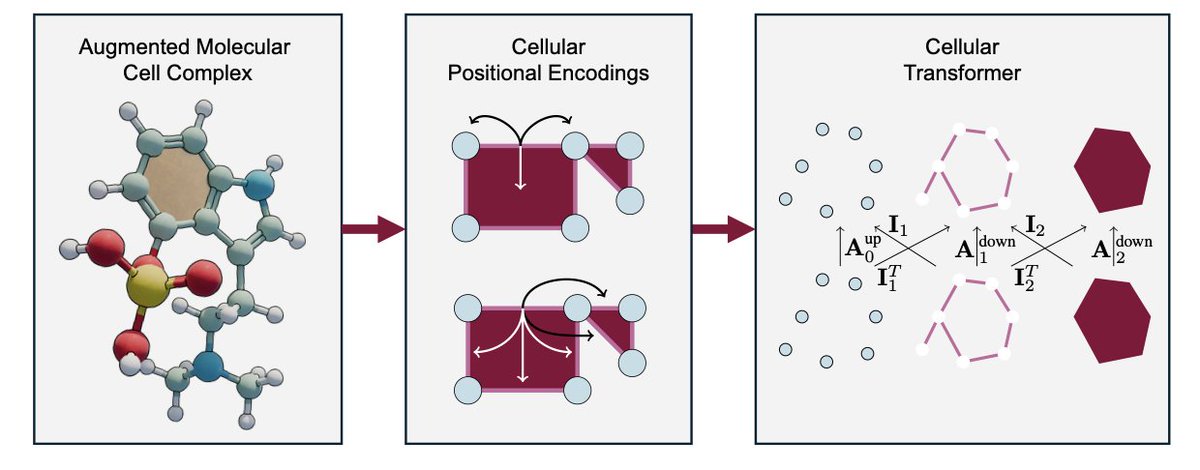
HIGHER-ORDER MOLECULAR LEARNING: THE CELLULAR TRANSFORMER
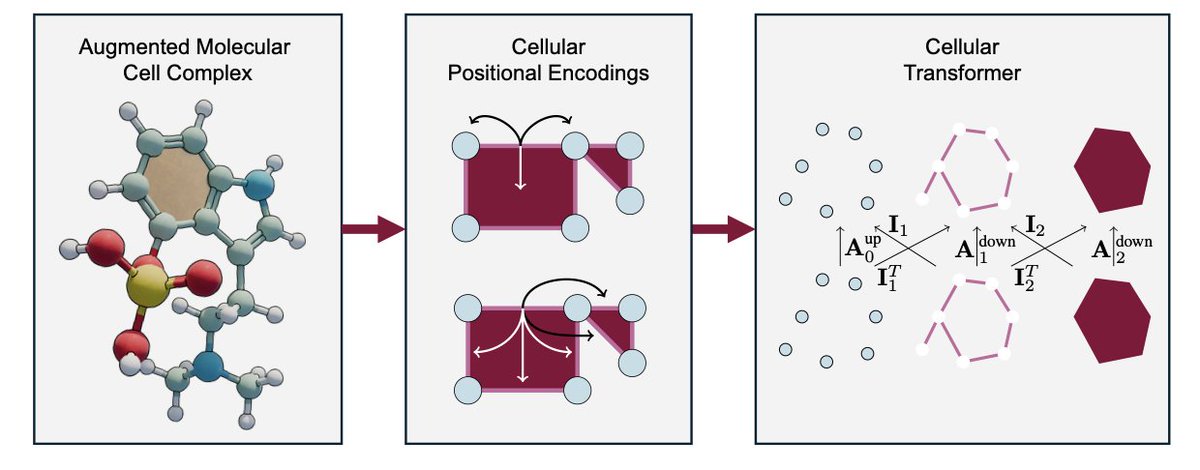
1.This paper introduces the Cellular Transformer (CT), a topological deep learning (TDL) framework that generalizes graph transformers to operate on cell complexes, enabling the modeling of higher-order molecular structures like rings, fused motifs, and multi-bond systems.
2.A key innovation is the augmented molecular cell complex (AMCC), a richer molecular representation where atoms, bonds, and rings are treated as 0-, 1-, and 2-cells, respectively—embedding chemical topology directly into the learning architecture.
3.CT performs attention not just over nodes or edges but across multiple structural ranks (0D, 1D, 2D) using a novel pairwise and general cellular attention mechanism, capturing multiscale interactions without relying on graph rewiring, virtual nodes, or ad-hoc biases.
4.The architecture employs tensor diagrams to formalize attention flow across cochain ranks, integrating both cross-rank and intra-rank attention, guided by neighborhood matrices derived from topological relations like incidence and adjacency.
5.To encode structure, CT introduces cellular positional encodings (CPEs), extending Laplacian and random walk encodings to the cellular domain. It also proposes a novel barycentric subdivision encoding (BSPe) that enhances topological locality.
6.Extensive benchmarking on MoleculeNet and the Graph Classification Benchmark (GCB) demonstrates that CT consistently outperforms GNNs, MPNNs, and graph transformers, especially in datasets where topological motifs matter most.
7.In GCB, CT achieved the highest accuracy (75.4%) compared to other message-passing and kernel-based methods, showing the benefit of high-order attention even in originally graph-based domains.
8.On MoleculeNet, CT ranked among the top across both classification (AUC) and regression (RMSE) tasks, performing particularly well in datasets like HIV, ClinTox, and ESOL, where higher-order features are vital.
9.The method is highly generalizable: lifting molecular graphs into CCs using tools like TopoX allows CT to apply broadly, even when only graph data is available, making it backward-compatible with existing pipelines.
10.This work positions CT as a foundation for topologically informed molecular modeling, offering a scalable, interpretable, and efficient alternative to current GNN-based methods, with applications across drug discovery and materials science.
📜Paper: openreview.net/pdf?id=GW3h79…
#MolecularModeling #TopologicalDeepLearning #GraphTransformer #DrugDiscovery #CellComplex #MoleculeNet #ICLR2025 #ChemicalML #AttentionMechanism #CellularTransformer
1
11
1,358
11 May 2025
HIGHER-ORDER MOLECULAR LEARNING: THE CELLULAR TRANSFORMER
1.This paper introduces the Cellular Transformer (CT), a topological deep learning (TDL) framework that generalizes graph transformers to operate on cell complexes, enabling the modeling of higher-order molecular structures like rings, fused motifs, and multi-bond systems.
2.A key innovation is the augmented molecular cell complex (AMCC), a richer molecular representation where atoms, bonds, and rings are treated as 0-, 1-, and 2-cells, respectively—embedding chemical topology directly into the learning architecture.
3.CT performs attention not just over nodes or edges but across multiple structural ranks (0D, 1D, 2D) using a novel pairwise and general cellular attention mechanism, capturing multiscale interactions without relying on graph rewiring, virtual nodes, or ad-hoc biases.
4.The architecture employs tensor diagrams to formalize attention flow across cochain ranks, integrating both cross-rank and intra-rank attention, guided by neighborhood matrices derived from topological relations like incidence and adjacency.
5.To encode structure, CT introduces cellular positional encodings (CPEs), extending Laplacian and random walk encodings to the cellular domain. It also proposes a novel barycentric subdivision encoding (BSPe) that enhances topological locality.
6.Extensive benchmarking on MoleculeNet and the Graph Classification Benchmark (GCB) demonstrates that CT consistently outperforms GNNs, MPNNs, and graph transformers, especially in datasets where topological motifs matter most.
7.In GCB, CT achieved the highest accuracy (75.4%) compared to other message-passing and kernel-based methods, showing the benefit of high-order attention even in originally graph-based domains.
8.On MoleculeNet, CT ranked among the top across both classification (AUC) and regression (RMSE) tasks, performing particularly well in datasets like HIV, ClinTox, and ESOL, where higher-order features are vital.
9.The method is highly generalizable: lifting molecular graphs into CCs using tools like TopoX allows CT to apply broadly, even when only graph data is available, making it backward-compatible with existing pipelines.
10.This work positions CT as a foundation for topologically informed molecular modeling, offering a scalable, interpretable, and efficient alternative to current GNN-based methods, with applications across drug discovery and materials science.
📜Paper: openreview.net/pdf?id=GW3h79…
#MolecularModeling #TopologicalDeepLearning #GraphTransformer #DrugDiscovery #CellComplex #MoleculeNet #ICLR2025 #ChemicalML #AttentionMechanism #CellularTransformer
2
822

3 May 2025
JTreeformer: Graph-Transformer via Latent-Diffusion Model for Molecular Generation
arxiv.org/abs/2504.20770
1/ 🔬 Introducing JTreeformer — a powerful new framework for molecular graph generation that merges graph transformers with latent diffusion models, achieving state-of-the-art results on MOSES and QM9 benchmarks. #AI #DrugDiscovery
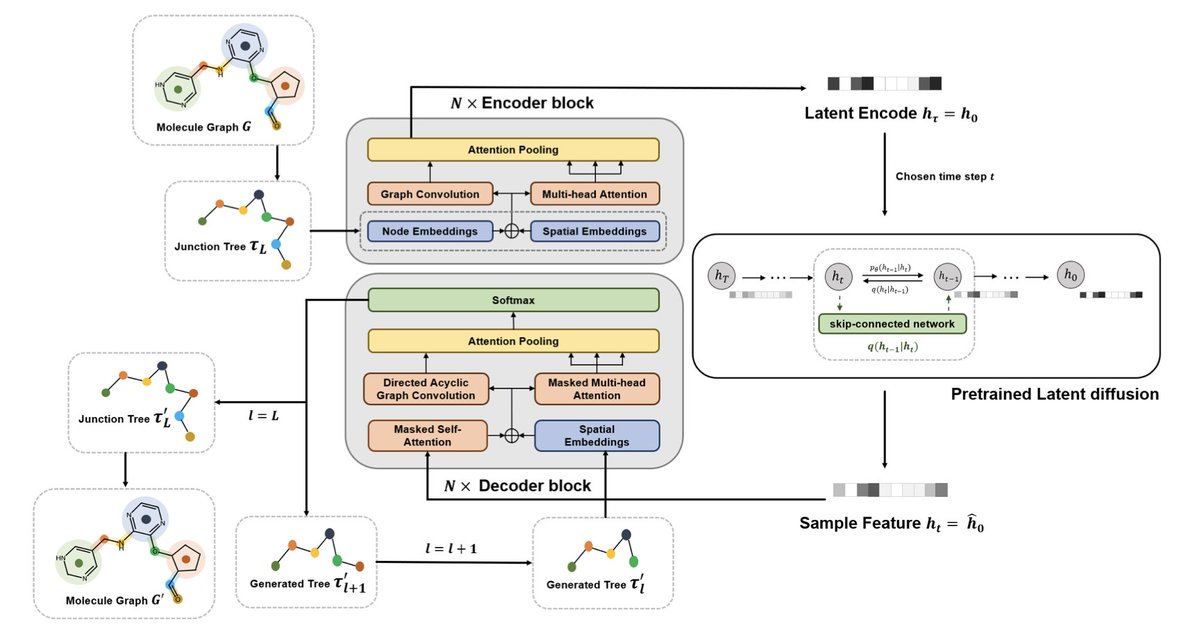
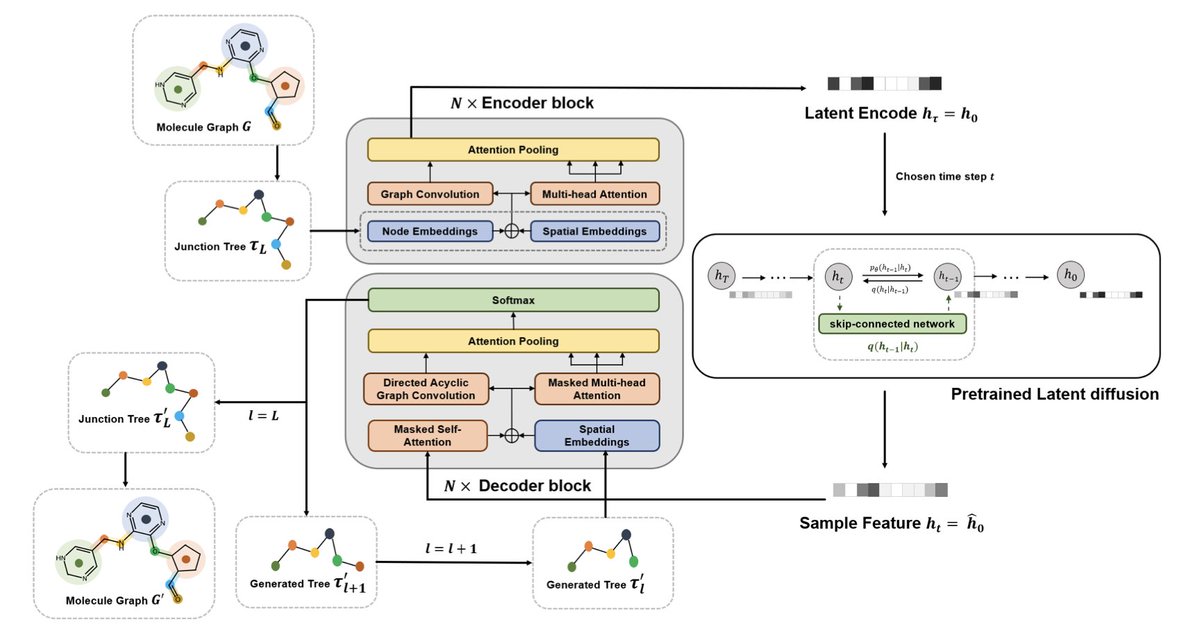
2/ 🧩 JTreeformer reframes molecule generation as a junction tree prediction task, enabling efficient, autoregressive generation with strong chemical validity and scaffold control.
3/ 🧠 The encoder blends Graph Convolutional Networks (GCNs) with multi-head self-attention, capturing both local atomic details and global structure across the tree.
4/ ➡️ In the decoder, a Directed Acyclic Graph Convolutional Network (DAGCN) masked attention allows sequential generation of molecular substructures—overcoming limitations of standard GCNs in autoregression.
5/ 🌫️ A latent diffusion model (DDIM) is used in the learned representation space for structured, controllable sampling—boosting diversity and validity of generated molecules.
6/ 🌲 Instead of neural discriminators (like in JT-VAE), JTreeformer uses Monte Carlo Tree Search (MCTS) to resolve isomer assemblies during the tree-to-graph step—faster and more efficient.
7/ 📊 JTreeformer outperforms JT-VAE, GraphAF, and LatentGAN on key metrics: validity, uniqueness, novelty, and internal diversity. It shines in scaffold-aware interpolation and latent clustering.
8/ 🧪 Ablation studies show removing DAGCN or junction tree features leads to major performance drops—both are essential to the model’s success.
9/ 🌀 Latent space visualizations reveal semantic clustering by molecular properties (logP, TPSA). Interpolations show smooth transitions between molecular scaffolds.
10/ 🧬 By uniting chemical grammar (junction trees), geometric learning (GCN/transformers), and generative modeling (diffusion), JTreeformer sets a new bar for controllable, interpretable molecular generation. 🌟
#GraphTransformer #DiffusionModels #DrugDiscovery #AIBiology #DeepLearning
3
131
1 May 2025
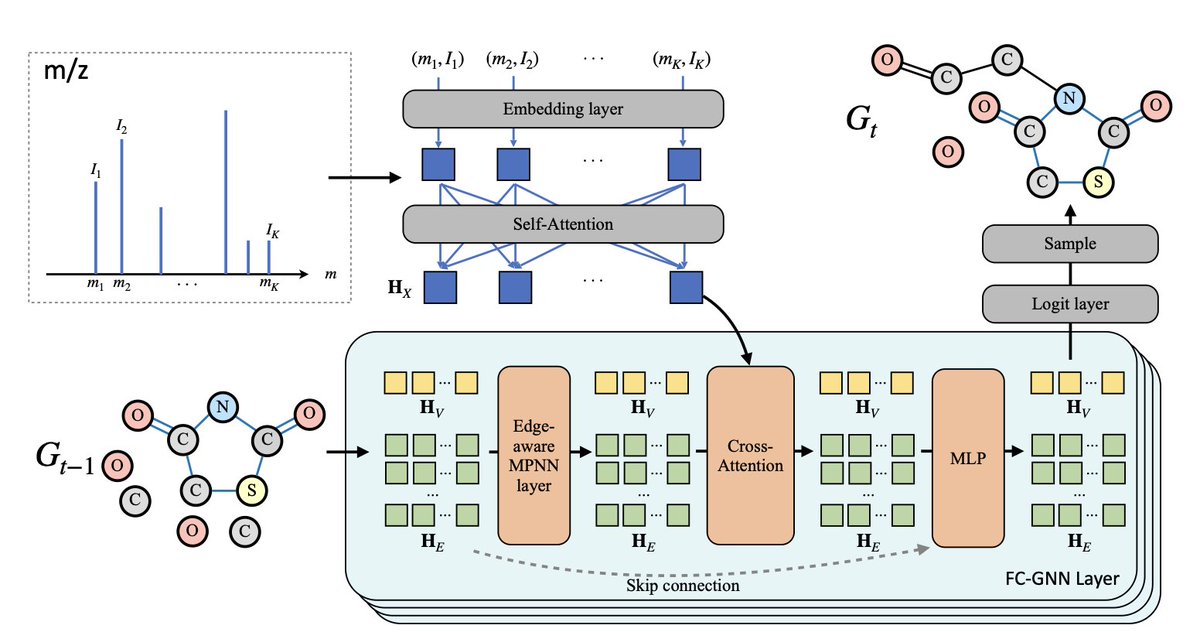
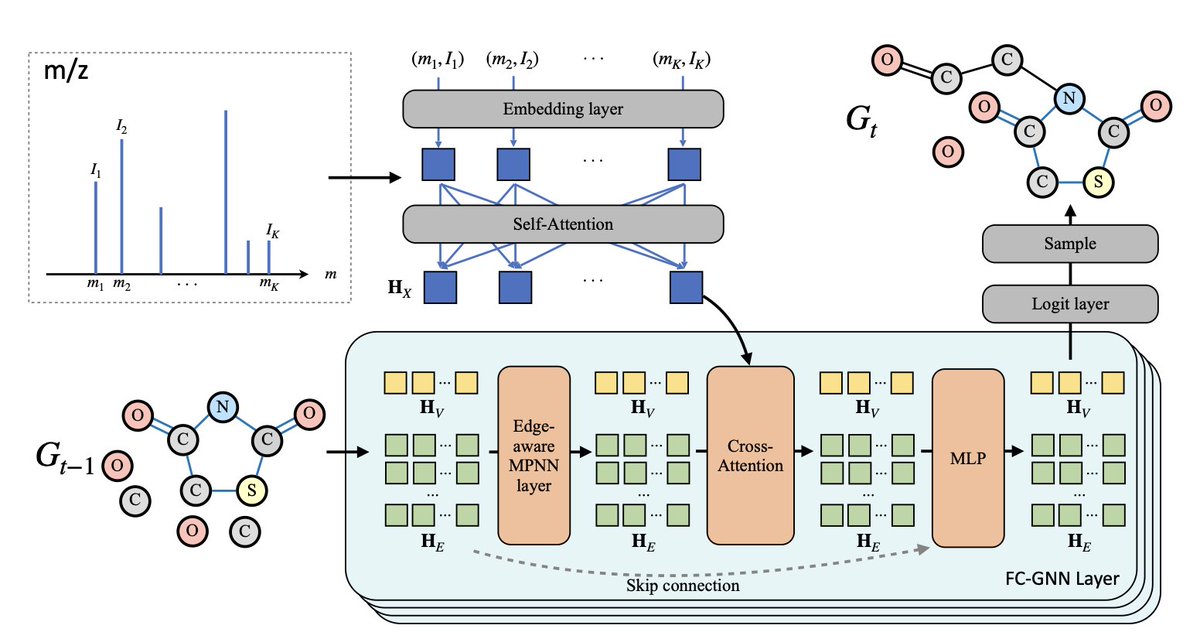
MADGEN: Mass-spec Attends to De Novo Molecular Generation
1. MADGEN introduces a two-stage deep learning framework for de novo molecular structure generation guided by mass spectrometry (MS/MS) data, aimed at illuminating the "dark chemical space" of unannotated spectra.
2. The first stage retrieves a molecular scaffold using contrastive learning to align mass spectra and scaffold representations in a shared latent space—framing scaffold retrieval as a ranking task based on spectrum-scaffold similarity.
3. Once a scaffold is retrieved, the second stage generates the full molecule by progressively adding atoms and bonds, guided by the MS/MS spectrum using a graph-based generative model inspired by a Markov bridge formulation.
4. Scaffold-guided generation reduces the search space and enhances interpretability, enabling the model to build upon a chemically plausible backbone instead of starting from scratch.
5. MADGEN integrates mass spectral data via self-attention and cross-attention in a graph transformer, and applies classifier-free guidance (CFG) to dynamically balance generation fidelity and diversity throughout the process.
6. On benchmark datasets NIST23, CANOPUS, and MassSpecGym, MADGEN outperforms prior methods like Spec2Mol, MSNovelist, and SMILES Transformers, particularly when scaffold retrieval is accurate.
7. Using oracle scaffolds, MADGEN achieves up to 49% top-1 accuracy and 0.80 Tanimoto similarity on NIST23—demonstrating the full potential of its scaffold-conditioned generation pipeline.
8. Ablation studies show that tokenization and self-attention for encoding spectral peaks, coupled with cross-attention on graph nodes and edges, significantly improve generation quality and molecular similarity.
9. Sensitivity analysis reveals that generation difficulty increases with the number of free atoms to connect, highlighting the benefit of larger or more informative scaffolds to simplify the generative task.
10. MADGEN offers a novel scaffold-centered approach to spectra-guided molecular generation, combining multimodal contrastive learning with structured graph generation—advancing the frontier of computational metabolomics.
💻Code: github.com/HassounLab/MADGEN
📜Paper: arxiv.org/abs/2501.01950
#MassSpectrometry #MolecularGeneration #ContrastiveLearning #GraphTransformer #DeNovoDesign #Metabolomics #AI4Science #ComputationalBiology #DeepLearning #ScaffoldDesign
2
5
948
1 May 2025
MADGEN: Mass-spec Attends to De Novo Molecular Generation
1. MADGEN introduces a two-stage deep learning framework for de novo molecular structure generation guided by mass spectrometry (MS/MS) data, aimed at illuminating the "dark chemical space" of unannotated spectra.
2. The first stage retrieves a molecular scaffold using contrastive learning to align mass spectra and scaffold representations in a shared latent space—framing scaffold retrieval as a ranking task based on spectrum-scaffold similarity.
3. Once a scaffold is retrieved, the second stage generates the full molecule by progressively adding atoms and bonds, guided by the MS/MS spectrum using a graph-based generative model inspired by a Markov bridge formulation.
4. Scaffold-guided generation reduces the search space and enhances interpretability, enabling the model to build upon a chemically plausible backbone instead of starting from scratch.
5. MADGEN integrates mass spectral data via self-attention and cross-attention in a graph transformer, and applies classifier-free guidance (CFG) to dynamically balance generation fidelity and diversity throughout the process.
6. On benchmark datasets NIST23, CANOPUS, and MassSpecGym, MADGEN outperforms prior methods like Spec2Mol, MSNovelist, and SMILES Transformers, particularly when scaffold retrieval is accurate.
7. Using oracle scaffolds, MADGEN achieves up to 49% top-1 accuracy and 0.80 Tanimoto similarity on NIST23—demonstrating the full potential of its scaffold-conditioned generation pipeline.
8. Ablation studies show that tokenization and self-attention for encoding spectral peaks, coupled with cross-attention on graph nodes and edges, significantly improve generation quality and molecular similarity.
9. Sensitivity analysis reveals that generation difficulty increases with the number of free atoms to connect, highlighting the benefit of larger or more informative scaffolds to simplify the generative task.
10. MADGEN offers a novel scaffold-centered approach to spectra-guided molecular generation, combining multimodal contrastive learning with structured graph generation—advancing the frontier of computational metabolomics.
💻Code: github.com/HassounLab/MADGEN
📜Paper: arxiv.org/abs/2501.01950
#MassSpectrometry #MolecularGeneration #ContrastiveLearning #GraphTransformer #DeNovoDesign #Metabolomics #AI4Science #ComputationalBiology #DeepLearning #ScaffoldDesign
4
746
30 Apr 2025
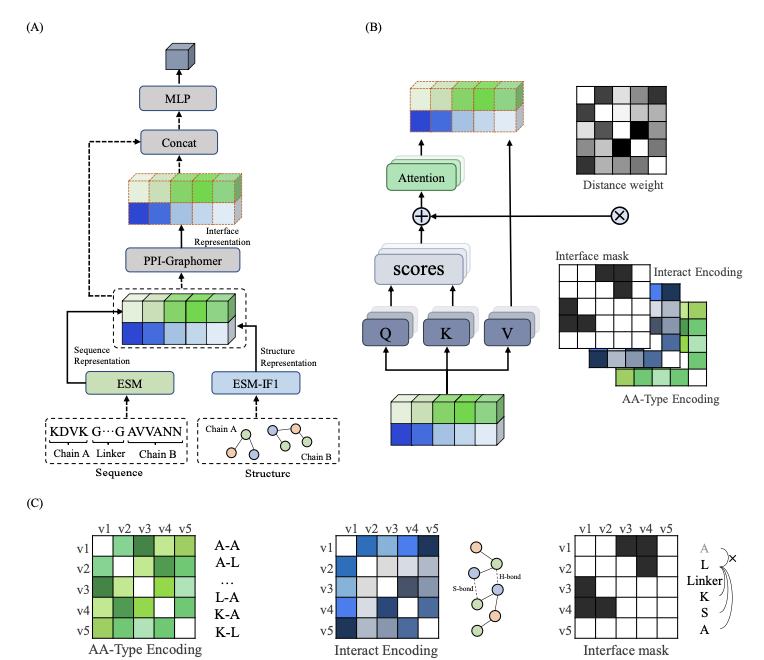
PPI-Graphomer: enhanced protein-protein affinity prediction using pretrained and graph transformer models
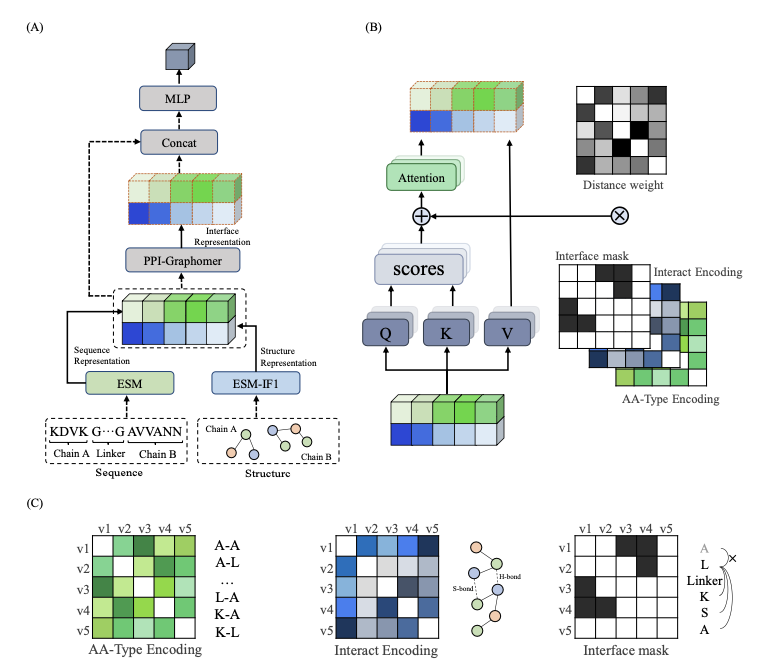
1. This study presents PPI-Graphomer, a deep learning framework that integrates pretrained protein language models with a graph transformer architecture to predict protein-protein binding affinities with high accuracy and interpretability.
2. The model leverages ESM2 for sequence representation and ESM-IF1 for structural features, combining these with a novel PPI-Graphomer module that explicitly models interface interactions between protein chains using chemical interaction encodings.
3. PPI-Graphomer applies attention bias terms based on amino acid pair types, interaction forces (e.g., hydrogen bonds, disulfide bonds), and spatial proximity within 7 Å, focusing attention on biologically meaningful interface residues.
4. A unique innovation is the encoding of interaction features directly into the self-attention mechanism of a graph-based transformer, allowing the model to capture inter-chain hotspot interactions crucial for accurate affinity prediction.
5. The model is trained and evaluated on benchmark datasets from PDBbind and achieves state-of-the-art performance, with PCC = 0.633 and MAE = 1.57 on a 162-sample test set, outperforming empirical methods like FoldX and Rosetta.
6. Compared to baseline models such as PRODIGY, CP-PIE, ISLAND, and PPI-Affinity, PPI-Graphomer consistently ranks first or second across multiple test sets, especially excelling on more diverse or complex samples.
7. Ablation studies show that removing either ESM2 features, ESM-IF1 features, or the PPI-Graphomer module leads to notable performance degradation—demonstrating that sequence, structure, and interface information are synergistic.
8. Notably, the sequence features from ESM2 contributed the most, likely due to the large-scale pretraining and higher-dimensional embeddings, but the addition of interface-specific encoding from PPI-Graphomer significantly boosts prediction precision.
9. On subsets of binary protein complexes, PPI-Graphomer achieved higher correlation scores than traditional scoring tools and even outperformed ColabFold’s iPAE metric, highlighting its robustness across structural formats.
10. By combining evolutionary-scale protein representations with interaction-aware attention mechanisms, PPI-Graphomer offers a powerful, extensible foundation for predictive modeling in PPI-driven drug discovery and structural biology.
💻Code: github.com/xiebaoshu058/PPI-…
📜Paper: bmcbioinformatics.biomedcent…
#ProteinInteractions #BindingAffinity #GraphTransformer #PretrainedModels #PPI #ComputationalBiology #AI4Science #Bioinformatics #ESM #StructurePrediction
4
684
30 Apr 2025
PPI-Graphomer: enhanced protein-protein affinity prediction using pretrained and graph transformer models
1. This study presents PPI-Graphomer, a deep learning framework that integrates pretrained protein language models with a graph transformer architecture to predict protein-protein binding affinities with high accuracy and interpretability.
2. The model leverages ESM2 for sequence representation and ESM-IF1 for structural features, combining these with a novel PPI-Graphomer module that explicitly models interface interactions between protein chains using chemical interaction encodings.
3. PPI-Graphomer applies attention bias terms based on amino acid pair types, interaction forces (e.g., hydrogen bonds, disulfide bonds), and spatial proximity within 7 Å, focusing attention on biologically meaningful interface residues.
4. A unique innovation is the encoding of interaction features directly into the self-attention mechanism of a graph-based transformer, allowing the model to capture inter-chain hotspot interactions crucial for accurate affinity prediction.
5. The model is trained and evaluated on benchmark datasets from PDBbind and achieves state-of-the-art performance, with PCC = 0.633 and MAE = 1.57 on a 162-sample test set, outperforming empirical methods like FoldX and Rosetta.
6. Compared to baseline models such as PRODIGY, CP-PIE, ISLAND, and PPI-Affinity, PPI-Graphomer consistently ranks first or second across multiple test sets, especially excelling on more diverse or complex samples.
7. Ablation studies show that removing either ESM2 features, ESM-IF1 features, or the PPI-Graphomer module leads to notable performance degradation—demonstrating that sequence, structure, and interface information are synergistic.
8. Notably, the sequence features from ESM2 contributed the most, likely due to the large-scale pretraining and higher-dimensional embeddings, but the addition of interface-specific encoding from PPI-Graphomer significantly boosts prediction precision.
9. On subsets of binary protein complexes, PPI-Graphomer achieved higher correlation scores than traditional scoring tools and even outperformed ColabFold’s iPAE metric, highlighting its robustness across structural formats.
10. By combining evolutionary-scale protein representations with interaction-aware attention mechanisms, PPI-Graphomer offers a powerful, extensible foundation for predictive modeling in PPI-driven drug discovery and structural biology.
💻Code: github.com/xiebaoshu058/PPI-…
📜Paper: bmcbioinformatics.biomedcent…
#ProteinInteractions #BindingAffinity #GraphTransformer #PretrainedModels #PPI #ComputationalBiology #AI4Science #Bioinformatics #ESM #StructurePrediction
5
26
1,794
30 Apr 2025
JTreeformer: Graph-Transformer via Latent-Diffusion Model for Molecular Generation
1. This study presents JTreeformer, a novel framework for molecular graph generation that integrates a graph transformer with latent diffusion modeling, achieving state-of-the-art results on MOSES and QM9 benchmarks.
2. JTreeformer transforms molecular generation into a junction tree generation task, enabling sequence-like modeling of molecular graphs with chemical validity and efficient autoregressive decoding.
3. The encoder combines Graph Convolutional Networks (GCNs) with multi-head self-attention to simultaneously capture local atomic context and global structural dependencies across the junction tree.
4. A Directed Acyclic Graph Convolutional Network (DAGCN) is introduced in the decoder, paired with masked attention to iteratively generate molecule fragments, resolving limitations of traditional GCNs in sequential prediction.
5. A key innovation is the use of a latent diffusion model (DDIM) in the learned representation space, allowing controlled and structured sampling that enhances the diversity and validity of generated molecules.
6. The model uses Monte Carlo Tree Search (MCTS) to resolve isomer assembly during tree-to-graph conversion, replacing costly neural discriminators used in prior works like JT-VAE.
7. JTreeformer outperforms leading baselines such as JT-VAE, GraphAF, and LatentGAN across validity, novelty, uniqueness, and internal diversity metrics—particularly excelling in scaffold-aware interpolation and latent space clustering.
8. Extensive ablation studies confirm the importance of both DAGCN and feature selection from the junction tree, showing substantial performance drops when either component is removed.
9. Visualization of the latent space reveals semantically meaningful clusters aligned with molecular properties (e.g., logP, TPSA), and interpolation experiments show smooth transitions between molecular scaffolds.
10. By uniting symbolic chemical grammar (junction trees), geometric learning (GCN/transformer), and generative sampling (diffusion), JTreeformer sets a new direction for robust, controllable, and interpretable molecular generation.
💻Code: anonymous.4open.science/r/JT…
📜Paper: arxiv.org/abs/2504.20770
#MolecularGeneration #GraphTransformer #DiffusionModels #DrugDiscovery #ComputationalBiology #AI4Science #Chemoinformatics #DeepLearning
1
8
42
4,791
30 Apr 2025
JTreeformer: Graph-Transformer via Latent-Diffusion Model for Molecular Generation
1. This study presents JTreeformer, a novel framework for molecular graph generation that integrates a graph transformer with latent diffusion modeling, achieving state-of-the-art results on MOSES and QM9 benchmarks.
2. JTreeformer transforms molecular generation into a junction tree generation task, enabling sequence-like modeling of molecular graphs with chemical validity and efficient autoregressive decoding.
3. The encoder combines Graph Convolutional Networks (GCNs) with multi-head self-attention to simultaneously capture local atomic context and global structural dependencies across the junction tree.
4. A Directed Acyclic Graph Convolutional Network (DAGCN) is introduced in the decoder, paired with masked attention to iteratively generate molecule fragments, resolving limitations of traditional GCNs in sequential prediction.
5. A key innovation is the use of a latent diffusion model (DDIM) in the learned representation space, allowing controlled and structured sampling that enhances the diversity and validity of generated molecules.
6. The model uses Monte Carlo Tree Search (MCTS) to resolve isomer assembly during tree-to-graph conversion, replacing costly neural discriminators used in prior works like JT-VAE.
7. JTreeformer outperforms leading baselines such as JT-VAE, GraphAF, and LatentGAN across validity, novelty, uniqueness, and internal diversity metrics—particularly excelling in scaffold-aware interpolation and latent space clustering.
8. Extensive ablation studies confirm the importance of both DAGCN and feature selection from the junction tree, showing substantial performance drops when either component is removed.
9. Visualization of the latent space reveals semantically meaningful clusters aligned with molecular properties (e.g., logP, TPSA), and interpolation experiments show smooth transitions between molecular scaffolds.
10. By uniting symbolic chemical grammar (junction trees), geometric learning (GCN/transformer), and generative sampling (diffusion), JTreeformer sets a new direction for robust, controllable, and interpretable molecular generation.
💻Code: anonymous.4open.science/r/JT…
📜Paper: arxiv.org/abs/2504.20770
#MolecularGeneration #GraphTransformer #DiffusionModels #DrugDiscovery #ComputationalBiology #AI4Science #Chemoinformatics #DeepLearning
1
14
1,414

7 Aug 2024
たんぱく質の立体構造から、GraphTransformerを使ってリガンドや薬剤の結合ポケットを予測するモデルの論文が、publishされました。🎉
journals.plos.org/plosone/ar…
1
22
139
11,319

👉 Learn how to build and query a knowledge graph from unstructured data using Diffbot API, FalkorDB, and LangChain in this awesome tutorial by @g_korland 🙌 #knowledgegraph #diffbot #falkordb #langchain #graphtransformer
falkordb.com/blog/diffbot-gr…
2
3
417

1 Nov 2021
The model is less powerful than 3-WL and more than 1-WL. But higher-order WL expressivity does not guarantee better generalization performance. We showed this previously: GNNs like GAT, GatedGCN or GraphTransformer outperform GIN and 3-WL GNN which have better WL expressivity.
2
4