
1 Dec 2025
.@BerkeleyLab's #AI experts are heading to San Diego this week for #NeurIPS 2025, joining the world’s leading minds in #MachineLearning & #datascience.
Check out our day-by-day participation guide and see how we’re shaping the future of #ML4Science: bit.ly/NeurIPS25_LBL

ALT Banner for Berkeley Lab at NeurIPS 2025, featuring the Neural Information Processing Systems logo and a photograph of San Diego’s downtown skyline at dusk, with city lights reflecting off the water.
1
1
9
935

22 Sep 2025
New Special Issue in Flow, Turbulence and Combustion! 🚀
“Machine Learning for Fluids” from PINNs & operator learning to flow control with RL (check our DRL papers there!!).
Editorial w/ Paola Cinnella: doi.org/10.1007/s10494-025-0…
#ML4Science #FluidDynamics

4
21
835

6 Aug 2025
🕊️ Early Bird ends Aug 15!
Join us at the 3rd Intl Symposium on Machine Learning in Quantum Chemistry (SMLQC), Oct 5–7.
Discounted accommodation now available!
Info: smlqc2025.com
#ML #QuantumChem #CompChem #ML4Chem #ChemInformatics #ChemML #ML4Science #MLInChemistry

1
8
670

1 Jul 2025
A Benchmark for Quantum Chemistry Relaxations via Machine Learning Interatomic Potentials
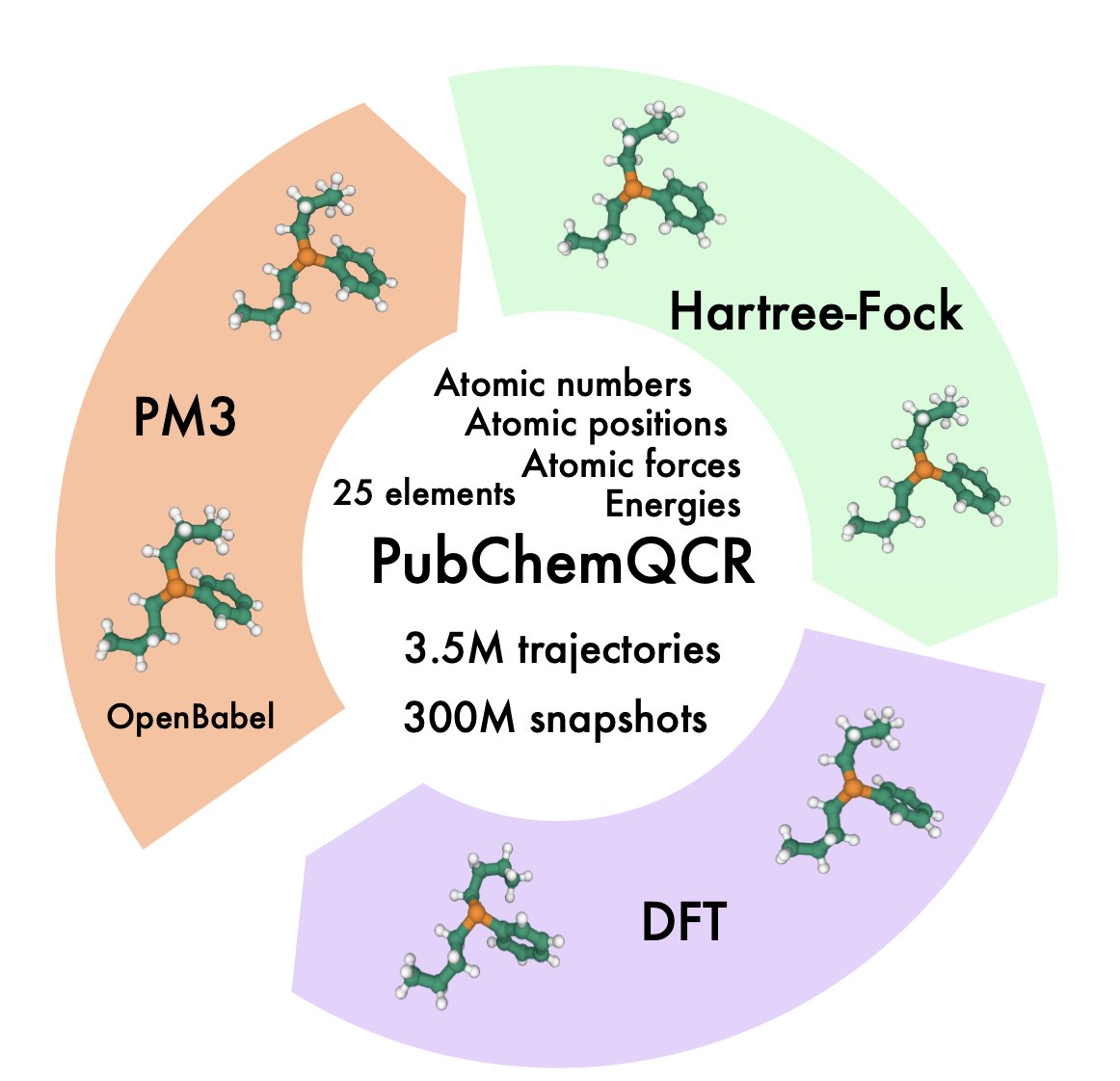
1.PubChemQCR is the largest publicly available dataset of DFT-based molecular relaxation trajectories, with 3.5 million molecules and over 300 million conformations, including 105 million computed with DFT. Each conformation includes total energy and atomic force labels.
2.The dataset captures full geometry optimization trajectories, not just final structures—addressing a key gap in previous datasets. This enables machine learning interatomic potentials (MLIPs) to learn from both stable and non-equilibrium geometries.
3.PubChemQCR offers broad chemical diversity, spanning 25 elements and a wide range of molecular sizes and conformational complexities. It was built from PubChemQC’s raw optimization outputs, spanning PM3, Hartree–Fock, and DFT stages.
4.Compared to existing datasets like QM9, GEOM, or ANI-1x, PubChemQCR provides significantly more conformational data, better element coverage, and crucial force labels at high-accuracy DFT level—making it uniquely suited for training MLIPs.
5.A curated subset, PubChemQCR-S, contains \~41K DFT relaxation trajectories for efficient model benchmarking. This subset supports rapid prototyping, ablation studies, and hyperparameter tuning.
6.The authors benchmarked 9 MLIP models (SchNet, PaiNN, NequIP, FAENet, Equiformer, etc.) on energy and force prediction tasks using PubChemQCR-S. Equiformer achieved the best overall performance on both energy and force metrics.
7.In geometry optimization tasks, Equiformer outperformed all other models, achieving 70.15% average energy minimization, 23.81% chemical accuracy success rate, and a 19.85% force convergence rate. Most other models struggled, especially with force convergence.
8.The dataset supports supervised pretraining of 3D molecular models with physically grounded energy and force labels—potentially benefiting downstream property prediction tasks in drug discovery and materials science.
9.It also enables training of generative models for 3D molecular structures. These models can learn to generate low-energy conformations directly from the data, bypassing costly DFT optimization.
10.Limitations include the dataset's near-equilibrium bias (due to DFT relaxation) and inconsistent label quality across optimization stages. Also, chemical element coverage is capped at 25 due to DFT method constraints.
11.Despite limitations, PubChemQCR is a foundational resource for building accurate, transferable, and data-efficient MLIPs. It can accelerate atomistic simulations, geometry optimization, and generative modeling in quantum chemistry.
💻Code: huggingface.co/divelab
📜Paper: arxiv.org/abs/2506.23008v1
#QuantumChemistry #ML4Science #DFT #GraphNeuralNetworks #MolecularSimulation #MachineLearning #OpenScience #MolecularModeling
1
6
612
1 Jul 2025
A Benchmark for Quantum Chemistry Relaxations via Machine Learning Interatomic Potentials
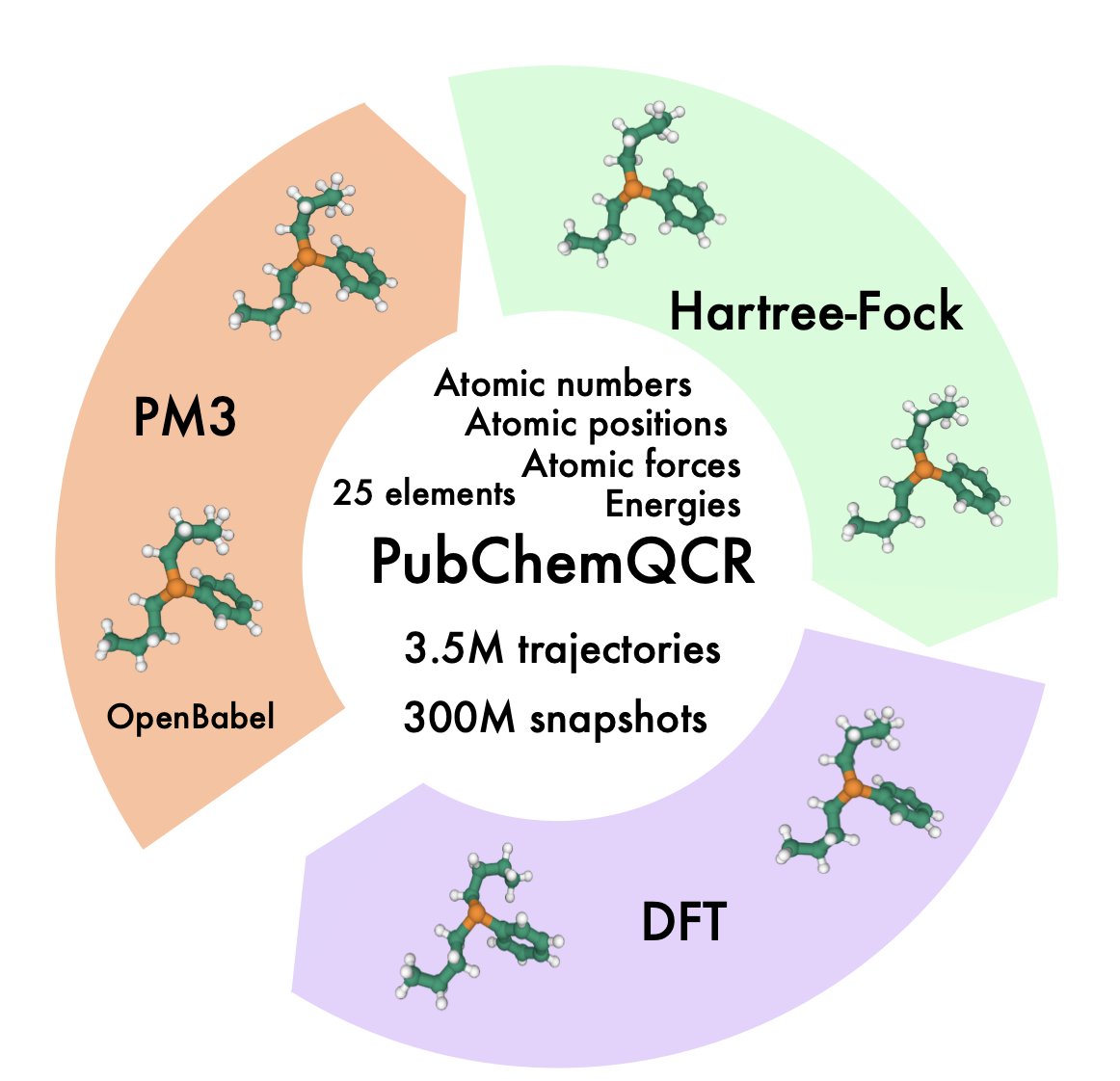
1.PubChemQCR is the largest publicly available dataset of DFT-based molecular relaxation trajectories, with 3.5 million molecules and over 300 million conformations, including 105 million computed with DFT. Each conformation includes total energy and atomic force labels.
2.The dataset captures full geometry optimization trajectories, not just final structures—addressing a key gap in previous datasets. This enables machine learning interatomic potentials (MLIPs) to learn from both stable and non-equilibrium geometries.
3.PubChemQCR offers broad chemical diversity, spanning 25 elements and a wide range of molecular sizes and conformational complexities. It was built from PubChemQC’s raw optimization outputs, spanning PM3, Hartree–Fock, and DFT stages.
4.Compared to existing datasets like QM9, GEOM, or ANI-1x, PubChemQCR provides significantly more conformational data, better element coverage, and crucial force labels at high-accuracy DFT level—making it uniquely suited for training MLIPs.
5.A curated subset, PubChemQCR-S, contains ~41K DFT relaxation trajectories for efficient model benchmarking. This subset supports rapid prototyping, ablation studies, and hyperparameter tuning.
6.The authors benchmarked 9 MLIP models (SchNet, PaiNN, NequIP, FAENet, Equiformer, etc.) on energy and force prediction tasks using PubChemQCR-S. Equiformer achieved the best overall performance on both energy and force metrics.
7.In geometry optimization tasks, Equiformer outperformed all other models, achieving 70.15% average energy minimization, 23.81% chemical accuracy success rate, and a 19.85% force convergence rate. Most other models struggled, especially with force convergence.
8.The dataset supports supervised pretraining of 3D molecular models with physically grounded energy and force labels—potentially benefiting downstream property prediction tasks in drug discovery and materials science.
9.It also enables training of generative models for 3D molecular structures. These models can learn to generate low-energy conformations directly from the data, bypassing costly DFT optimization.
10.Limitations include the dataset's near-equilibrium bias (due to DFT relaxation) and inconsistent label quality across optimization stages. Also, chemical element coverage is capped at 25 due to DFT method constraints.
11.Despite limitations, PubChemQCR is a foundational resource for building accurate, transferable, and data-efficient MLIPs. It can accelerate atomistic simulations, geometry optimization, and generative modeling in quantum chemistry.
💻Code: huggingface.co/divelab
📜Paper: arxiv.org/abs/2506.23008v1
#QuantumChemistry #ML4Science #DFT #GraphNeuralNetworks #MolecularSimulation #MachineLearning #OpenScience #MolecularModeling
12
44
2,082
29 Jun 2025
Mapping the space of protein binding sites with sequence-based protein language models
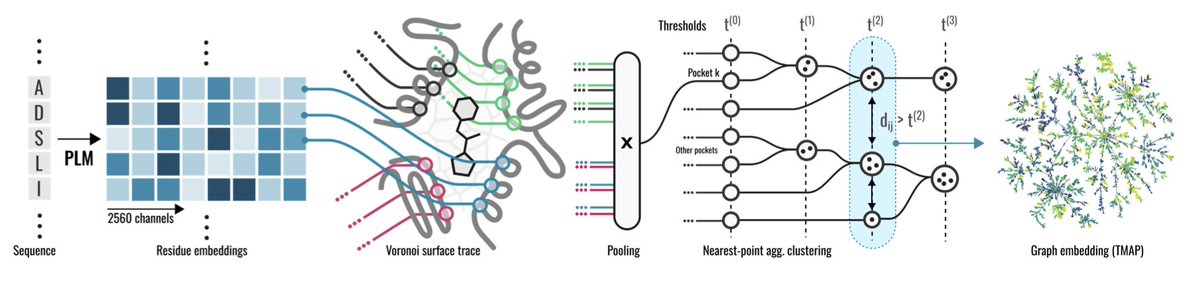
1.This paper introduces EPoCS, an alignment-free method for representing and comparing protein binding sites using embeddings from protein language models combined with 3D structural information.
2.EPoCS leverages ESM-2 embeddings mapped onto pocket interface residues identified via 3D tessellation around bound ligands, capturing functional, structural, and evolutionary aspects of binding sites.
3.The resulting vector representations support robust Euclidean distance metrics for pocket similarity, enabling meaningful hierarchical clustering and efficient navigation of the "pocketome".
4.Compared to traditional metrics like APoc or sequence similarity, EPoCS strikes a multiscale balance between local structure and global sequence features, with better correlation in similarity-onset regions.
5.Using a 96k-pocket dataset from the PDB, the authors demonstrate that EPoCS clusters pockets meaningfully—capturing cases where proteins with low sequence identity but structurally similar pockets are grouped together.
6.Visualised as a "Pocket Atlas" using TMAP, EPoCS clusters reflect both local biochemical coherence (e.g., similar ligands or active sites) and broader functional relationships (e.g., shared EC classes), while avoiding overfitting to global sequence.
7.EPoCS can identify data leakage in benchmark datasets. For example, it reveals instances in PoseBusters where low-sequence-identity validation sets still overlap with training structures via pocket similarity, undermining benchmark reliability.
8.The method enables principled, adjustable debiasing of machine learning train-test splits via cluster- or tree-based separation in pocket similarity space. This allows generation of successively harder validation scenarios for ML models.
9.Druggability prediction using ESM-2 embeddings (via a simple MLP model) confirms the effectiveness of EPoCS-based splits: random splits yield inflated AUCs (~0.92), while harder EPoCS splits reveal true generalisation performance (~0.81).
10.EPoCS is computationally cheap and alignment-free, outperforming alignment-based methods like APoc in scalability while preserving biologically meaningful structure.
11.The authors suggest EPoCS could be extended beyond ligand-bound pockets to orphan sites, and integrated into pipelines for ligand transfer, fragment mapping, and cryptic pocket detection.
12.By offering a unifying, sequence-based yet structure-aware similarity metric, EPoCS sets the stage for more trustworthy benchmarking and improved model selection in structure-based drug discovery.
💻Code: github.com/tugceoruc/epocs
📜Paper: academic.oup.com/bioinformat…
#ProteinDesign #StructureBasedDrugDesign #ML4Science #PocketSimilarity #ProteinLanguageModel #EPoCS #Bioinformatics

1
6
37
2,573
29 Jun 2025
Mapping the space of protein binding sites with sequence-based protein language models
1.This paper introduces EPoCS, an alignment-free method for representing and comparing protein binding sites using embeddings from protein language models combined with 3D structural information.
2.EPoCS leverages ESM-2 embeddings mapped onto pocket interface residues identified via 3D tessellation around bound ligands, capturing functional, structural, and evolutionary aspects of binding sites.
3.The resulting vector representations support robust Euclidean distance metrics for pocket similarity, enabling meaningful hierarchical clustering and efficient navigation of the "pocketome".
4.Compared to traditional metrics like APoc or sequence similarity, EPoCS strikes a multiscale balance between local structure and global sequence features, with better correlation in similarity-onset regions.
5.Using a 96k-pocket dataset from the PDB, the authors demonstrate that EPoCS clusters pockets meaningfully—capturing cases where proteins with low sequence identity but structurally similar pockets are grouped together.
6.Visualised as a "Pocket Atlas" using TMAP, EPoCS clusters reflect both local biochemical coherence (e.g., similar ligands or active sites) and broader functional relationships (e.g., shared EC classes), while avoiding overfitting to global sequence.
7.EPoCS can identify data leakage in benchmark datasets. For example, it reveals instances in PoseBusters where low-sequence-identity validation sets still overlap with training structures via pocket similarity, undermining benchmark reliability.
8.The method enables principled, adjustable debiasing of machine learning train-test splits via cluster- or tree-based separation in pocket similarity space. This allows generation of successively harder validation scenarios for ML models.
9.Druggability prediction using ESM-2 embeddings (via a simple MLP model) confirms the effectiveness of EPoCS-based splits: random splits yield inflated AUCs (~0.92), while harder EPoCS splits reveal true generalisation performance (~0.81).
10.EPoCS is computationally cheap and alignment-free, outperforming alignment-based methods like APoc in scalability while preserving biologically meaningful structure.
11.The authors suggest EPoCS could be extended beyond ligand-bound pockets to orphan sites, and integrated into pipelines for ligand transfer, fragment mapping, and cryptic pocket detection.
12.By offering a unifying, sequence-based yet structure-aware similarity metric, EPoCS sets the stage for more trustworthy benchmarking and improved model selection in structure-based drug discovery.
💻Code:
github.com/tugceoruc/epocs
📜Paper:
academic.oup.com/bioinformat…
#ProteinDesign #StructureBasedDrugDesign #ML4Science #PocketSimilarity #ProteinLanguageModel #EPoCS #Bioinformatics
1
22
92
4,985

19 Jun 2025
🚨PhD Opportunity in Computational Catalysis!
Join our team at @CICenergiGUNE to advance atomistic modelling & AI for sustainable nitrogen conversion 🌱
See details below.
#PhDposition #ComputationalChemistry #Catalysis #DFT #ML4Science @Chemjobber @TCD_Chemistry @TCD_Energy
1
8
18
1,508

27 May 2025
🚨 New article out now & featured on the cover of JCTC!
We introduce a hybrid ML/MM method using ANI-predicted charges for on-the-fly electrostatics in multiscale simulations
👉 doi.org/10.1021/acs.jctc.4c0…
@ACSPublications
#ML4Science #JCTC

1
2
3
74
13 May 2025
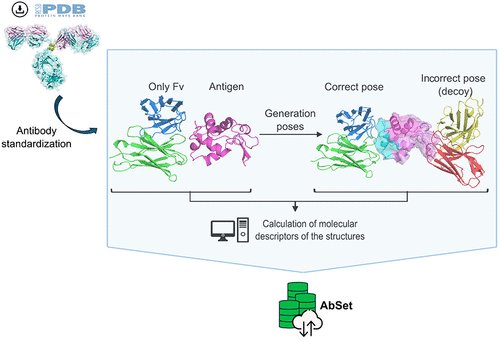
AbSet: A Standardized Data Set of Antibody Structures for Machine Learning Applications @JCIM_JCTC
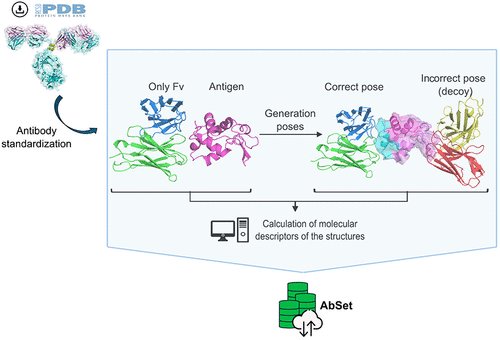
1.AbSet presents the most comprehensive and standardized structural dataset of antibodies to date—over 800,000 structures—designed to advance ML in therapeutic antibody discovery.
2.Unlike prior databases, AbSet includes both experimentally determined and in silico generated antibody–antigen complexes, annotated with molecular descriptors at the residue level.
3.The dataset integrates extensive docking simulations to generate high, medium, acceptable, and incorrect binding poses, enabling robust training and evaluation of AI models, including decoy-based learning.
4.AbSet employs a rigorous structure standardization pipeline using ANARCI-based numbering and antibody-specific filtering, capturing both paired and single-chain antibodies across formats like VH-VL, scFv, and VHH.
5.More than 14,000 PDB files are curated, with ~9,100 structures involving antigens and ~5,000 being free antibodies. Antigen types span a wide biological spectrum, with ~21% targeting SARS-CoV-2 spike proteins.
6.In silico structures were generated by docking modeled antibodies (via ABodyBuilder2) against crystal antigens using HADDOCK2.4, yielding nearly a million binding poses with diverse interaction quality.
7.Every in silico complex is scored using DockQ, providing quantitative benchmarks for pose accuracy and enabling separation of correct and incorrect binding predictions in training tasks.
8.AbSet also offers per-residue molecular descriptors—such as solvent-accessible area, depth, hydrophobicity, torsion angles, and secondary structure—making it ready-to-use for machine learning applications.
9.Unlike tools like AbNum or SAbDab, AbSet provides better standardization and includes recent entries, covering hundreds of antibody structures not annotated in other public databases.
10.Descriptors and structure files are accessible through Zenodo and GitHub, along with tools for filtering and processing the data efficiently, allowing scalable integration into ML workflows.
11.This work directly addresses the scarcity of high-quality structural antibody datasets, especially those that include antigen context and fine-grained annotations, positioning AbSet as a vital resource.
12.AbSet is particularly valuable for structure-based ML models in immunoinformatics, antibody docking, paratope prediction, and decoy-based benchmarking.
💻Code: github.com/SFBBGroup/AbSet
📜Paper: pubs.acs.org/doi/10.1021/acs…
#AntibodyDesign #MachineLearning #StructuralBioinformatics #ProteinDocking #DrugDiscovery #Immunoinformatics #Bioinformatics #AntibodyEngineering #OpenData #CDR #ML4Science
17
85
10,489
13 May 2025
AbSet: A Standardized Data Set of Antibody Structures for Machine Learning Applications @JCIM_JCTC
1.AbSet presents the most comprehensive and standardized structural dataset of antibodies to date—over 800,000 structures—designed to advance ML in therapeutic antibody discovery.
2.Unlike prior databases, AbSet includes both experimentally determined and in silico generated antibody–antigen complexes, annotated with molecular descriptors at the residue level.
3.The dataset integrates extensive docking simulations to generate high, medium, acceptable, and incorrect binding poses, enabling robust training and evaluation of AI models, including decoy-based learning.
4.AbSet employs a rigorous structure standardization pipeline using ANARCI-based numbering and antibody-specific filtering, capturing both paired and single-chain antibodies across formats like VH-VL, scFv, and VHH.
5.More than 14,000 PDB files are curated, with ~9,100 structures involving antigens and ~5,000 being free antibodies. Antigen types span a wide biological spectrum, with ~21% targeting SARS-CoV-2 spike proteins.
6.In silico structures were generated by docking modeled antibodies (via ABodyBuilder2) against crystal antigens using HADDOCK2.4, yielding nearly a million binding poses with diverse interaction quality.
7.Every in silico complex is scored using DockQ, providing quantitative benchmarks for pose accuracy and enabling separation of correct and incorrect binding predictions in training tasks.
8.AbSet also offers per-residue molecular descriptors—such as solvent-accessible area, depth, hydrophobicity, torsion angles, and secondary structure—making it ready-to-use for machine learning applications.
9.Unlike tools like AbNum or SAbDab, AbSet provides better standardization and includes recent entries, covering hundreds of antibody structures not annotated in other public databases.
10.Descriptors and structure files are accessible through Zenodo and GitHub, along with tools for filtering and processing the data efficiently, allowing scalable integration into ML workflows.
11.This work directly addresses the scarcity of high-quality structural antibody datasets, especially those that include antigen context and fine-grained annotations, positioning AbSet as a vital resource.
12.AbSet is particularly valuable for structure-based ML models in immunoinformatics, antibody docking, paratope prediction, and decoy-based benchmarking.
💻Code: github.com/SFBBGroup/AbSet
📜Paper: pubs.acs.org/doi/10.1021/acs…
#AntibodyDesign #MachineLearning #StructuralBioinformatics #ProteinDocking #DrugDiscovery #Immunoinformatics #Bioinformatics #AntibodyEngineering #OpenData #CDR #ML4Science
3
6
1,119
11 May 2025
Assessing Quantization and Efficient Fine-Tuning for Protein Language Models
1.This paper provides the first comprehensive benchmark of applying quantization and parameter-efficient fine-tuning (QLoRA) to protein language models (PLMs), enabling 4-bit training and inference with minimal performance loss across diverse protein tasks.
2.By quantizing PLMs like ESM-2, ESM-C, ProtBERT, and ProtT5, the authors achieve up to 90% memory reduction—dropping memory needs from 60 GB to under 10 GB—while retaining over 90% of predictive performance on fluorescence, stability, and secondary structure tasks.
3.QLoRA-based models match or even outperform full models in some settings; notably, ESM-C 600M achieved near-parity on all tasks with 50% less memory, and surpassed the much larger ESM-2 3B in stability prediction.
4.This makes large-scale protein modeling accessible to academic labs and smaller institutions, as quantized models can now run on consumer-grade GPUs and free cloud platforms like Google Colab.
5.The study evaluates compute metrics like memory footprint and power usage across models and batch sizes, showing consistent reductions—on average 46.7% less memory and 5.6% lower power consumption.
6.On the generative side, ProtGPT2 and ProLLaMA models were also 4-bit quantized for unconditional protein generation, with no significant degradation in FoldSeek scores, pLDDT, secondary structure content, or disorder prediction.
7.Predicted proteins from quantized models retained structure-aware metrics, such as alignment scores and predicted template modeling (pTM), showing their viability for use in structure-based generative design.
8.The authors observed that quantization is most effective at smaller batch sizes; at large batch sizes, memory reduction diminishes or even reverses, aligning with known scaling behaviors of activation gradients.
9.The paper also emphasizes the growing importance of computational accessibility: with PLMs approaching the size of general-purpose LLMs, democratizing their usage via quantization and LoRA is critical for innovation in protein science.
10.With support for fine-tuning and generation across multiple architectures, the QLoRA protocol offers a universal, low-barrier approach for training PLMs in diverse biological settings.
11.Overall, this study shows that 4-bit quantization plus LoRA adaptation enables scalable and affordable protein modeling at negligible cost to performance—paving the way for broader adoption of PLMs in life science research.
📜Paper: openreview.net/forum?id=KBMx…
#ProteinLanguageModels #Quantization #LoRA #EfficientAI #Bioinformatics #ProteinDesign #PLMs #ICLR2025 #QLoRA #ESM #ProtGPT2 #ProLLaMA #ML4Science
9
33
2,992
6 May 2025
BOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
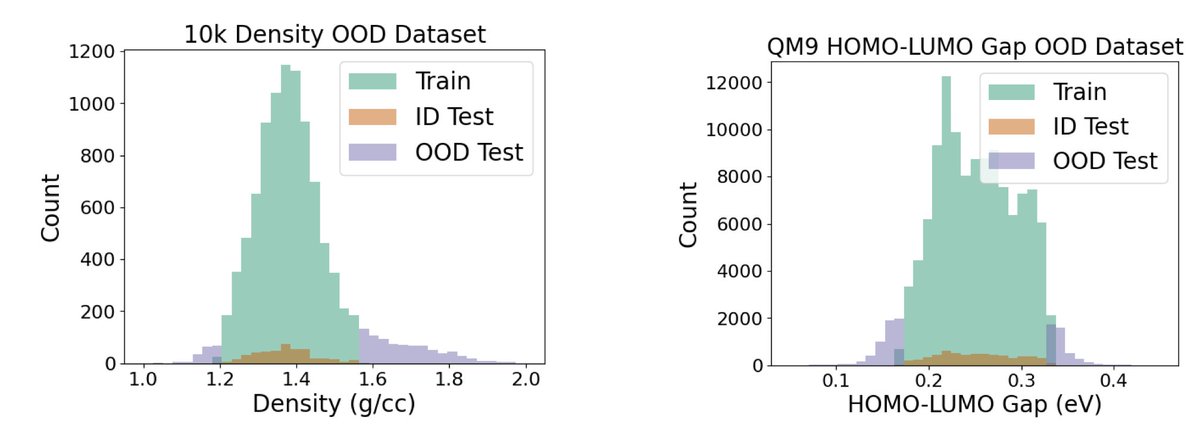
1. BOOM is the first systematic benchmark focused on evaluating the out-of-distribution (OOD) generalization performance of molecular property prediction models, a critical challenge for enabling ML-guided discovery of novel molecules.
2. The authors assess 12 ML models across 10 molecular properties using over 140 model-task combinations, revealing that even top models exhibit up to 3x higher error on OOD data compared to in-distribution (ID) test sets.
3. No current model shows robust OOD performance across all tasks. MACE leads in OOD performance on 5 of 10 tasks, while ET dominates ID tasks, indicating that strong in-distribution accuracy does not guarantee generalization.
4. Pretraining strategies like masked language modeling (MLM) significantly improve ID performance but fail to enhance, and sometimes degrade, OOD performance—highlighting a key limitation in current chemical foundation models.
5. The benchmark defines OOD splits based on the tails of molecular property distributions, aligning with real-world discovery goals where desirable molecules often lie outside known distributions.
6. 3D-aware models, especially those with E(3)-equivariance like EGNN and MACE, outperform SMILES-based transformers in OOD settings. Representation choice is thus more critical than scale for extrapolation.
7. Hyperparameter tuning targeting OOD performance offers some benefit, particularly for simple properties like density or heat of formation, but is not sufficient to close the generalization gap.
8. Data augmentation by including a small number of OOD molecules in training substantially improves generalization for 7 of 8 tasks tested, suggesting that even modest exposure to rare examples helps overcome distributional shifts.
9. ModernBERT, though a transformer model, incorporates architecture changes that improve OOD performance in tasks like HoF and Cv, narrowing the gap with graph-based models and showing promise for LLM-style scalability.
10. The study identifies specific property types (e.g., dipole moment, HOMO, LUMO) as persistent weak points for OOD prediction, likely due to the absence of explicit electronic structure features in most models.
11. BOOM provides an open-source benchmark, dataset, and codebase to standardize OOD evaluation and accelerate the development of chemically generalizable machine learning models.
12. This work positions OOD generalization—not just ID accuracy—as a new frontier for chemical ML, essential for reliable property extrapolation and robust molecular discovery.
📜Paper: arxiv.org/abs/2505.01912
#Chemoinformatics #OutOfDistribution #MachineLearning #MolecularDesign #GraphNeuralNetworks #MolecularProperty #Benchmarking #ML4Science #GNN #SMILES #Pretraining #DataAugmentation #BOOM

4
15
1,210
6 May 2025
BOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
1. BOOM is the first systematic benchmark focused on evaluating the out-of-distribution (OOD) generalization performance of molecular property prediction models, a critical challenge for enabling ML-guided discovery of novel molecules.
2. The authors assess 12 ML models across 10 molecular properties using over 140 model-task combinations, revealing that even top models exhibit up to 3x higher error on OOD data compared to in-distribution (ID) test sets.
3. No current model shows robust OOD performance across all tasks. MACE leads in OOD performance on 5 of 10 tasks, while ET dominates ID tasks, indicating that strong in-distribution accuracy does not guarantee generalization.
4. Pretraining strategies like masked language modeling (MLM) significantly improve ID performance but fail to enhance, and sometimes degrade, OOD performance—highlighting a key limitation in current chemical foundation models.
5. The benchmark defines OOD splits based on the tails of molecular property distributions, aligning with real-world discovery goals where desirable molecules often lie outside known distributions.
6. 3D-aware models, especially those with E(3)-equivariance like EGNN and MACE, outperform SMILES-based transformers in OOD settings. Representation choice is thus more critical than scale for extrapolation.
7. Hyperparameter tuning targeting OOD performance offers some benefit, particularly for simple properties like density or heat of formation, but is not sufficient to close the generalization gap.
8. Data augmentation by including a small number of OOD molecules in training substantially improves generalization for 7 of 8 tasks tested, suggesting that even modest exposure to rare examples helps overcome distributional shifts.
9. ModernBERT, though a transformer model, incorporates architecture changes that improve OOD performance in tasks like HoF and Cv, narrowing the gap with graph-based models and showing promise for LLM-style scalability.
10. The study identifies specific property types (e.g., dipole moment, HOMO, LUMO) as persistent weak points for OOD prediction, likely due to the absence of explicit electronic structure features in most models.
11. BOOM provides an open-source benchmark, dataset, and codebase to standardize OOD evaluation and accelerate the development of chemically generalizable machine learning models.
12. This work positions OOD generalization—not just ID accuracy—as a new frontier for chemical ML, essential for reliable property extrapolation and robust molecular discovery.
📜Paper: arxiv.org/abs/2505.01912
#Chemoinformatics #OutOfDistribution #MachineLearning #MolecularDesign #GraphNeuralNetworks #MolecularProperty #Benchmarking #ML4Science #GNN #SMILES #Pretraining #DataAugmentation #BOOM
8
999
2 May 2025
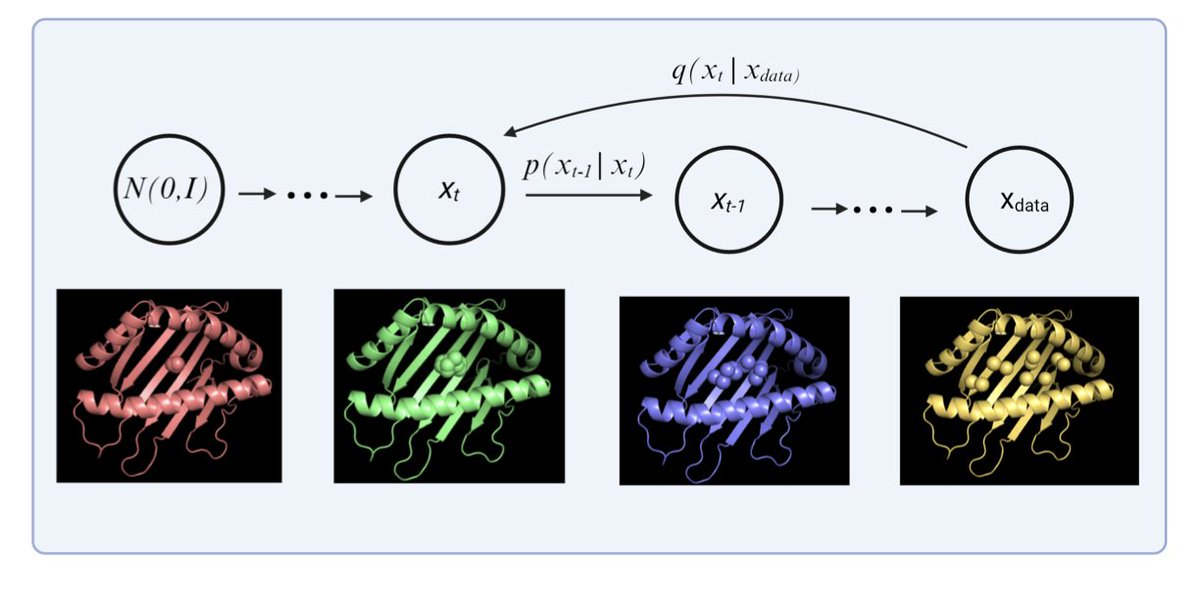
Fast and Accurate Peptide–MHC Structure Prediction via an Equivariant Diffusion Model
1. MHC-Diff introduces a new paradigm for modeling peptide–MHC structures using a SE(3)-equivariant diffusion model, enabling sub-angstrom accuracy in C-alpha atom prediction while supporting diverse peptide lengths and MHC alleles.
2. Unlike conventional models that output a single static conformation, MHC-Diff generates multiple diverse structures, capturing the inherent flexibility of peptide binding and significantly improving the odds of recovering native-like conformations.
3. On the 8K benchmark (HLA-A\*02:01, 9-mers), MHC-Diff achieves a best-of-10 RMSD of 0.45 Å and average RMSD of 0.99 Å, outperforming AlphaFold2-FineTune, Pandora, and Ape-Gen 2.0 by a large margin.
4. MHC-Diff generalizes well to unseen alleles and longer peptides in the 100K benchmark, achieving 0.61 Å best-of-10 RMSD and maintaining structural fidelity across complex input diversity.
5. Its efficiency is unmatched—generating 10 diverse pMHC structures in just 5.6 seconds—while preserving high accuracy, making it well-suited for large-scale screening and downstream applications like immunogenicity prediction.
6. By employing E(3)-equivariant graph neural networks and chain-aware positional encodings, MHC-Diff respects geometric symmetries and learns conformational distributions rather than deterministic structures.
7. The model explicitly models structural flexibility—a critical factor for understanding T-cell receptor interactions and immune recognition—while remaining computationally efficient.
8. MHC-Diff’s open-source implementation and curated datasets are released publicly, offering the community a powerful tool for structure-based immunotherapy development.
💻Code: github.com/DavidFruehbuss/MH…
📜Paper: biorxiv.org/content/10.1101/…
#Immunotherapy #ProteinStructure #DiffusionModels #GeometricDeepLearning #CancerVaccine #pMHC #ML4Science #StructuralBiology #DeepLearning
5
35
4,223

🌍 AI is transforming climate research! Catch us at #EGU25 presenting cutting-edge work on applying #MachineLearning, hybrid models, and causal discovery in climate research.
⚡️ Don’t miss: Camps-Valls, Reid, Ouala, Beucler more!
#AI4Climate #ML4Science #EGU25 #ClimateAI

2
65

12 Mar 2025
Very happy to announce that I'll be joining the University of Tübingen (@uni_tue) as a Full Professor (W3) of "Machine Learning for Science" in the Computer Science Department within the Faculty of Science and the #ExcellenceCluster ML4Science.
Tübingen is an exceptionally vibrant place for AI/ML research with many many great groups and initiatives, and with strong applications in the natural sciences. Couldn't be happier; can't wait to start and meet all future colleagues, and building new and more powerful AI tools for understanding the universe.
We have numerous open positions (also within my @ERC_Research StG), for post-docs and PhDs. If you are excited to build AI-driven discovery of physics experiments for new ways to observe the universe (super-fast physics simulators, AI-driven exploration of large complex spaces, etc) and agent-based idea generations, send me an e-Mail.
20
5
145
9,362

Ready to start with #CEBRA for your behavioral clustering, neural data, or joint-modeling? Check out our new demo notebook to help you learn the best practices and how to develop models! cebra.ai/docs/demo_notebooks…
🌈🦓 #ML4Science
14
42
3,669

11 Feb 2025
It was a great experience to be part of the ML4Science 2025 conference in Shillong. Met some amazing people and felt even more motivated to work harder. 👨🏭

7
192

7 Feb 2025
Early morning Run walk on Shillong’s hilly roads with @joshiphy; we are here for the ML4Science symposium.
Covered three touristic places by 9AM. Quite productive, huh 😎.


3
12
798