Flexible Kernels for Protein Property Prediction
1. The paper introduces LOCK-GP: Gaussian processes with a new protein sequence kernel that combines evolutionary substitution matrices (e.g., BLOSUM) with an explicit “local linearity” inductive bias to model protein property landscapes from sparse experimental data.
2. Key kernel idea (LOCK: Locally Linear Correlation Kernel): replace one-hot “same/different” comparisons with amino-acid similarity from substitution matrices, and learn landscape-specific Hadamard-power exponents to tune how strongly similarities are amplified/attenuated while preserving kernel validity.
3. A central technical observation: many BLOSUM matrices are not only PSD but also infinitely divisible, so elementwise exponentiation by any positive power preserves PSD. This enables learnable exponents inside the GP kernel without breaking positive semidefiniteness.
4. LOCK is built from (i) an additive “linear” correlation kernel and (ii) a multiplicative “RBF-like” correlation kernel, then combined so predictions are nuanced and non-linear near training data but revert to a robust linear predictor farther away (avoiding both aggressive linear extrapolation and mean-reversion to the prior mean).
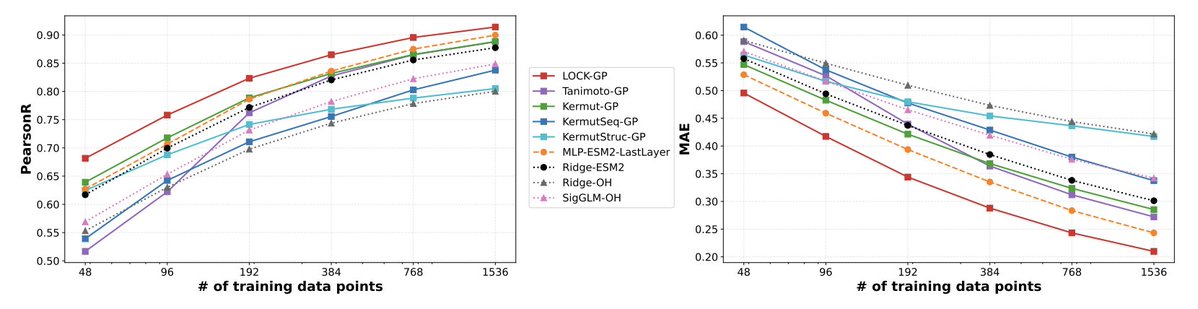
5. Benchmarking is extensive: 30 predictors evaluated across 21 protein property datasets (thermostability, binding affinity, fluorescence, capsid viability, etc.) under three regimes: i.i.d. CV, Hamming-distance extrapolation, and an “unseen mutations” OOD regime where test sequences include mutations absent from training.
6. Results highlight data efficiency and uncertainty quality: across datasets and training sizes (e.g., 48–1536 points), LOCK-GP is typically best or near-best on correlation and error metrics, and shows strong calibration via proper scoring rules like CRPS; uncertainty improves notably when local linearity is included.
7. A notable empirical takeaway: a sequence-only LOCK-GP that relies on a small substitution-matrix prior can frequently outperform or match baselines that depend on large foundation models (e.g., ESM-2 embeddings, structure features, ProteinMPNN-derived features), especially in extrapolation and OOD “unseen mutation” settings where high-dimensional embeddings can be fragile.
8. The paper generalizes LOCK to CLOCK (structure-conditioned LOCK): positional structure embeddings from a foundation model are mapped to position-specific amino-acid correlation matrices (parameterized as exp(-||z_a - z_a'||^2)), effectively learning structure-aware substitution behavior that can be used “zero-shot” as a kernel prior and then refined by GP training.
9. Multi-task learning: CLOCK-GP is trained across 371 thermostability landscapes (Tsuboyama et al.), showing that learning a shared, structure-conditioned kernel across landscapes yields strong performance; CLOCK-GP is especially competitive in low-landscape regimes (e.g., training on 10 landscapes), and learned correlations are interpretable (e.g., proline preferences near helix N-termini; arginine favored on surfaces vs cores).
10. Additional demonstrations: LOCK-GP supports GP-based Bayesian optimization via Thompson sampling to control exploration/diversity in design, and extends to binary classification (e.g., quantized fluorescence) with strong accuracy scaling with dataset size.
💻Code: github.com/generatebio/lock_…
📜Paper: arxiv.org/abs/2606.11057
#ComputationalBiology #ProteinEngineering #GaussianProcesses #MachineLearning #Kernels #ProteinDesign #UncertaintyQuantification #MultiTaskLearning #FoundationModels #Bioinformatics
2
11
1,110

May 27
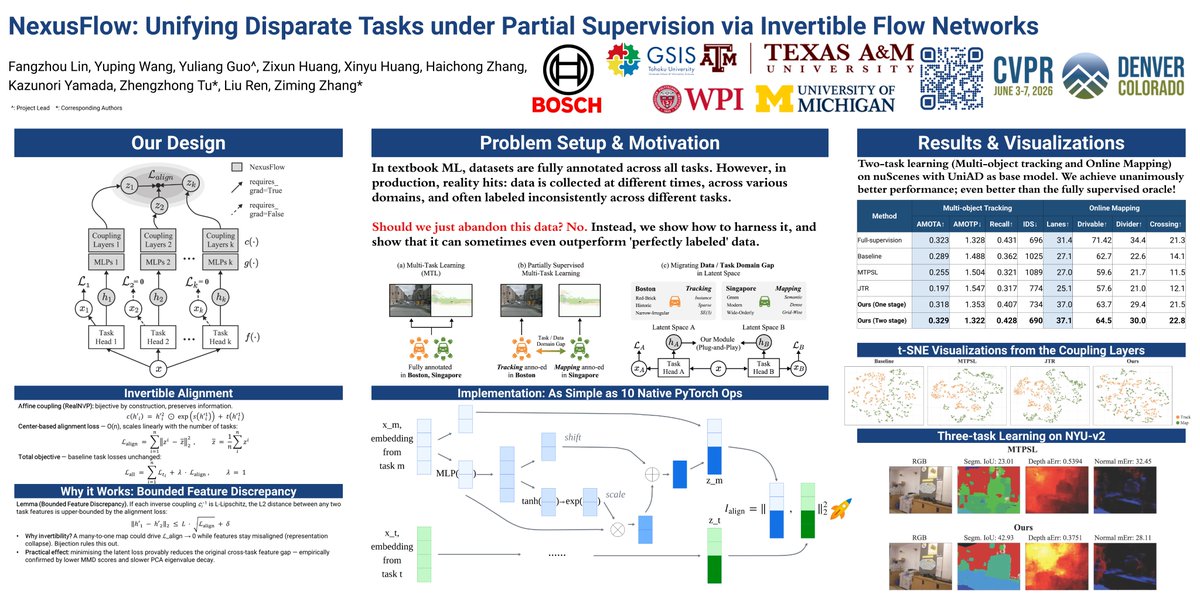
🎉 Excited to share our new work accepted to #CVPR2026 “𝗡𝗲𝘅𝘂𝘀𝗙𝗹𝗼𝘄: 𝗨𝗻𝗶𝗳𝘆𝗶𝗻𝗴 𝗗𝗶𝘀𝗽𝗮𝗿𝗮𝘁𝗲 𝗧𝗮𝘀𝗸𝘀 𝘂𝗻𝗱𝗲𝗿 𝗣𝗮𝗿𝘁𝗶𝗮𝗹 𝗦𝘂𝗽𝗲𝗿𝘃𝗶𝘀𝗶𝗼𝗻 𝘃𝗶𝗮 𝗜𝗻𝘃𝗲𝗿𝘁𝗶𝗯𝗹𝗲 𝗙𝗹𝗼𝘄 𝗡𝗲𝘁𝘄𝗼𝗿𝗸𝘀”
In textbooks and benchmarks, datasets are often neatly annotated for every task. In the real world, they rarely are. Data is collected at different times, in different places, and for different purposes. One dataset may contain labels for mapping, another for tracking, another for depth or segmentation. Does that mean fragmented data has to be discarded?
💪 𝗢𝘂𝗿 𝗮𝗻𝘀𝘄𝗲𝗿: 𝗻𝗼. We show that partially supervised, heterogeneous data can still be highly valuable—and in some cases, can even outperform fully annotated data.
How do we learn across structurally different tasks when labels are only partially available?
💡 𝗢𝘂𝗿 𝗦𝗼𝗹𝘂𝘁𝗶𝗼𝗻: 𝗡𝗲𝘅𝘂𝘀𝗙𝗹𝗼𝘄
NexusFlow is a lightweight, plug-and-play framework that aligns disparate tasks in a shared latent space.
What makes it work:
• 🔄 𝗜𝗻𝘃𝗲𝗿𝘁𝗶𝗯𝗹𝗲 𝗳𝗲𝗮𝘁𝘂𝗿𝗲 𝗮𝗹𝗶𝗴𝗻𝗺𝗲𝗻𝘁. Invertible coupling layers map task features into a unified canonical space. Since the mapping is bijective, task information is preserved, helping avoid the representational collapse often seen in vanilla alignment methods.
• 🔌 𝗣𝗹𝘂𝗴-𝗮𝗻𝗱-𝗽𝗹𝗮𝘆 𝗱𝗲𝘀𝗶𝗴𝗻. No need to modify task heads or losses. NexusFlow can be added to BEV-based backbones with a simple alignment loss.
• 📈 𝗦𝗰𝗮𝗹𝗮𝗯𝗹𝗲 𝘁𝗼 𝗺𝘂𝗹𝘁𝗶𝗽𝗹𝗲 𝘁𝗮𝘀𝗸𝘀. The method scales as O(N) with one surrogate branch per task, making extension to 3 tasks straightforward.
• 📐 𝗧𝗵𝗲𝗼𝗿𝗲𝘁𝗶𝗰𝗮𝗹 𝗴𝗿𝗼𝘂𝗻𝗱𝗶𝗻𝗴. Invertibility provides a provable bound that connects the alignment loss to cross-task knowledge transfer.
🏆 𝗥𝗲𝘀𝘂𝗹𝘁𝘀
NexusFlow sets a new state of the art on nuScenes for domain-partitioned autonomous driving, where online map reconstruction and multi-object tracking are supervised in different geographic regions.
It also delivers consistent gains across all three NYUv2 tasks: semantic segmentation, depth estimation, and surface normal prediction.
📎 𝗣𝗿𝗼𝗷𝗲𝗰𝘁 𝗽𝗮𝗴𝗲: ark1234.github.io/nexusflow_…
🤝 This work was conducted in collaboration across Worcester Polytechnic Institute, Texas A&M University, Tohoku University, University of Michigan, and Bosch Research.
Huge thanks to collaborators: Fangzhou Lin, Yuping Wang, Yuliang Guo, Zixun Huang, Xinyu Huang, Haichong Zhang, Kazunori Yamada, Zhengzhong Tu, Liu Ren, and Ziming Zhang.
#CVPR2026 #ComputerVision #MultiTaskLearning #AI #GenAI #AutonomousDriving #DeepLearning #RepresentationLearning
1
18
2,130
Multi-task gradient boosting with multi-modal molecular representations for simultaneous prediction of drug clearance and volume of distribution
1. The paper presents MTGBM, a multi-task gradient boosting machine that predicts two physiologically coupled PK endpoints at once: human clearance (CL) and volume of distribution (VD), using shared decision trees rather than separate single-task models.
2. A key design choice is multi-modal input: (i) CNN embeddings from 2D molecular structure images (ResNet18-based), (ii) MLP embeddings from concatenated RDKit descriptors Morgan fingerprints (256 bits) Mol2Vec (300d), and (iii) physicochemical in vitro preclinical PK variables (e.g., pKa, fup, rat/dog/monkey PK).
3. Both CNN and MLP pipelines are trained per target (CL-specific and VD-specific), then compressed into compact 2D bottleneck embeddings; these embeddings are later used as features for the boosting models, enabling the final learner to combine learned representations with experimental/nonclinical variables.
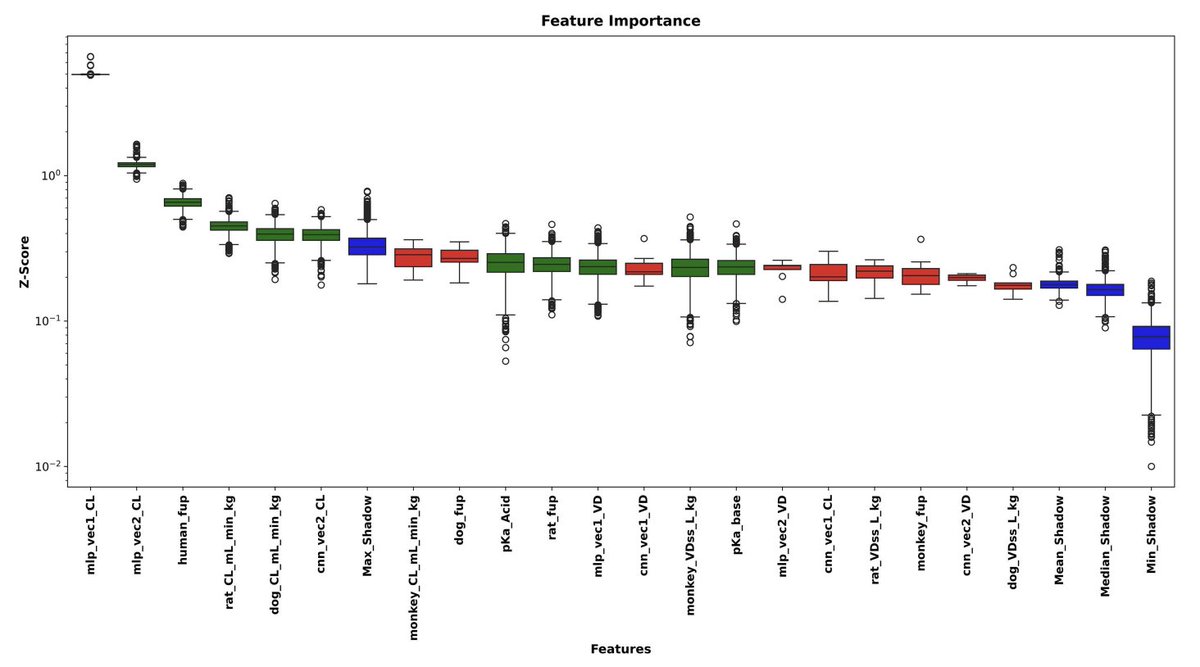
4. Feature selection is handled with BorutaSHAP (5,000 iterations, LightGBM backbone). It selects 11 features for CL and 11 for VD, with 6 overlapping; the multi-task model uses the union set (16 total), mixing modalities rather than relying on a single feature type.
5. On a held-out test set (n=139), MTGBM improves MSE and R² for both targets versus single-task LightGBM: CL MSE 19.081 → 14.580 (−23.6%), R² 0.140 → 0.196; VD MSE 1.130 → 0.757 (−33.0%), R² 0.212 → 0.418.
6. The paper highlights a metric tradeoff for VD: despite better MSE/R², overall VD GMFE worsens (2.014 → 2.286). A stratified analysis shows the degradation is driven specifically by the low-VD region (<0.5 L/kg), while mid and high VD ranges show improved GMFE under MTGBM.
7. Statistical testing with the Diebold–Mariano test supports the MSE improvements as significant for both endpoints (CL p=0.019; VD p=0.004), framing the gains as unlikely due to chance under their evaluation setup.
8. Robustness is assessed via 10 repeated random 60/20/20 splits with the full pipeline rerun each time (normalization, embedding training, BorutaSHAP, LightGBM tuning, MTGBM training). MTGBM wins on test MSE in 8/10 runs for CL and 10/10 runs for VD, suggesting consistent directional benefit, especially for VD.
9. SHAP interpretability suggests complementary modality contributions and cross-target signal usage: CL is strongly influenced by MLP embeddings, and VD is dominated by preclinical PK (notably dog_VDss_L_kg), while embeddings trained for one target can appear among top predictors for the other—consistent with CL/VD physiological coupling, but also influenced by the supervised nature of the embeddings.
📜Paper: doi.org/10.1371/journal.pone…
#ComputationalPharmacokinetics #ADME #MachineLearning #MultiTaskLearning #LightGBM #DrugDiscovery #QSAR #ModelInterpretability #SHAP #Pharmacometrics
1
7
1,062
When does context help? A systematic study of target-conditional molecular property prediction
1. The paper presents a systematic map of when target context helps (and hurts) molecular property prediction, spanning 10 protein families, 4 context-fusion designs, training data regimes from 67–9,409 compounds per target, and both random vs temporal splits.
2. Central result: how context is fused dominates whether context helps at all. With the same target identity signal, FiLM conditioning beats concatenation by 24.2 AUC points and beats additive-only conditioning by 8.6 points—showing that naive “add context” can be worse than no context.
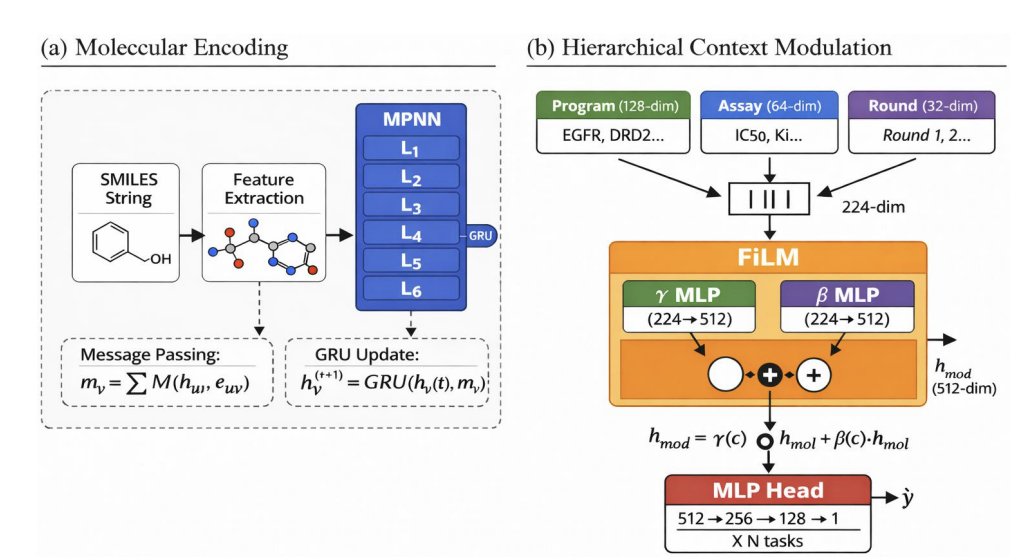
3. The model, NESTDRUG, uses an MPNN molecular encoder plus hierarchical context embeddings (target/program L1, assay L2, temporal round L3) and applies FiLM modulation: hmod = γ(c) ⊙ hmol β(c). The multiplicative γ term accounts for most of FiLM’s gain by selectively amplifying/suppressing molecular features per target.
4. In controlled ablations, target-specific L1 embeddings improve 9/10 DUD-E targets (mean 5.7 AUC points, p < 0.01). The largest gains are on ESR1 ( 13.4) and EGFR ( 13.2), suggesting context mainly helps by adapting to target-specific data/assay idiosyncrasies.
5. The clearest “context enables otherwise impossible prediction” case is CYP3A4, where only 67 training actives are available at the chosen activity threshold. A per-target Random Forest collapses to 0.238 AUC, while multi-task transfer with NESTDRUG reaches 0.686 AUC, indicating context-conditioned multitask learning can rescue data-scarce targets.
6. Context is not universally beneficial: BACE1 degrades by 10.2 AUC points with correct L1, attributed to distribution mismatch between ChEMBL (e.g., peptidomimetic series) and DUD-E (different scaffold distribution). The paper also reports few-shot adaptation of L1 embeddings consistently underperforms zero-shot (generic L1), warning against “quick embedding tuning” for new targets.
7. Mechanistic analysis suggests FiLM learns biologically structured modulation: kinase contexts yield γ > 1 (amplifying certain heterocycle/H-bond acceptor features), GPCR contexts yield γ < 1 (shifting emphasis toward lipophilicity). Inter-family variance in FiLM parameters exceeds intra-family variance (p < 0.001).
8. The work also audits benchmarking pitfalls on DUD-E: 1-nearest-neighbor Tanimoto similarity reaches 0.991 mean AUC without learning, and ~50% of actives overlap with ChEMBL training (highly target-dependent). The takeaway is that absolute DUD-E performance can be misleading, and leakage/structural bias can dominate apparent gains.
9. To address benchmark artifacts, the paper reports a temporal split evaluation (train ≤2020, test 2021–2024) with stable performance (overall 0.843 ROC-AUC, no year-over-year degradation), arguing this provides more realistic evidence that context-conditional representations can generalize to future chemical space.
💻Code: github.com/bryanc5864/nest-d…
📜Paper: arxiv.org/abs/2604.06558
#ComputationalBiology #Cheminformatics #DrugDiscovery #GraphNeuralNetworks #MachineLearning #ICLR #VirtualScreening #MultitaskLearning #DistributionShift #Benchmarking
3
10
1,249
Pan–Pharmacological Drug–Target Interaction Prediction with 3D–Informed Protein Encoding at Scale
1 OmniBind is a multitask DTI framework that predicts four pharmacological endpoints at once (pKd, pKi, pIC50, pEC50) from a single compound–protein pair, aiming to output a “pan-pharmacological profile” rather than optimizing for only one assay type.
2 The key scaling idea is to bring protein 3D information into sequence-speed inference: protein tertiary structure is converted into a 1D 3Di token sequence (20-letter structural alphabet) generated from sequence-derived structure, then modeled with Transformers like a language sequence.
3 Proteins are encoded in two parallel modalities: (a) amino-acid sequence Transformer encoder (captures evolutionary/sequence cues) and (b) 3Di-structure Transformer encoder (captures local biophysical environments). A learnable gated fusion layer dynamically weights each modality per input, instead of static addition/concatenation.
4 On the compound side, OmniBind uses a molecular graph representation from SMILES with a 1-layer GCN (chosen to avoid over-smoothing). A “virtual atom” connected to all atoms is added to aggregate global context, following prior CPI Transformer designs.
5 Compound–protein interaction is modeled by a 5-layer Transformer decoder: self-attention over compound atoms plus cross-attention to the fused protein representation. The virtual-atom embedding is used to emit four endpoint predictions in parallel.
6 Scale and data realism: trained on BindingDB May 2023 after extensive cleaning and unit harmonization (all endpoints converted to pActivity = −log10(M)). The final training snapshot contains 2,282,997 compound–protein pairs; temporal benchmarking uses newly added records in BindingDB Nov 2024.
7 Ablations show why gating matters: sequence-only and 3Di-only perform similarly; naive combination (element-wise addition) fails to consistently improve; gated fusion is best across regression and classification metrics, supporting the claim that sequence and 3Di provide complementary signals that must be context-weighted.
8 Generalization is tested with two protocols designed to penalize shortcut learning: (a) label reversal test (adversarial ligand-bias probe) and (b) temporal validation where both compounds and proteins are unseen (train up to May 2023; test on new Nov 2024 entries). OmniBind outperforms DTI-LM and TransformerCPI2.0 in both settings, suggesting it relies less on ligand–protein co-occurrence patterns.
9 Interpretability: cross-attention analysis on ABL1–imatinib highlights the known gatekeeper residue T315 (mapped to residue 93 in the kinase-domain construct). Introducing the resistance mutation T315I reduces attention at that position, indicating sensitivity to clinically relevant single-residue changes.
10 Practical demonstrations: proteome-wide screening against 20,421 reviewed human proteins recovers clozapine’s known clinical target landscape strongly (6/7 known therapeutic/off-targets within top 200; HTR2A ranked #1), and distinguishes clozapine vs clomipramine despite shared tricyclic scaffolds (HTR2A vs SLC6A4 as top-ranked primary targets). A separate repositioning workflow screens 1,615 FDA-approved drugs against PDE5, KLKB1, SIRT3 and uses Boltz-2 MOE docking for structural plausibility checks, yielding avanafil (PDE5) as an external positive control and proposing glecaprevir (KLKB1) and valrubicin (SIRT3) as candidates.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #DrugDiscovery #DTI #DeepLearning #Transformers #ProteinStructure #MultitaskLearning #BindingAffinity #Cheminformatics

2
23
1,518

16 Dec 2025
HeMeNet: Heterogeneous Multichannel Equivariant Network for Protein Multi-task Learning
1. Researchers propose a novel multi-task learning model called HeMeNet, which integrates six biologically relevant tasks, including affinity prediction and property prediction, into a single framework. This approach leverages shared knowledge across tasks to improve model performance and generalization.
2. HeMeNet introduces a heterogeneous full-atom graph representation that captures detailed interactions between different atoms in proteins. This is a significant innovation as it allows the model to handle various types of protein structures, from single chains to complexes, in a unified manner.
3. The model incorporates an E(3)-equivariant architecture, ensuring that predictions remain consistent under rotations, reflections, and translations of the input protein structures. This geometric invariance is crucial for accurate biological predictions.
4. A task-aware readout mechanism is developed to enable the model to generate task-specific outputs. This feature allows HeMeNet to perform harmonious multi-task training, avoiding interference between tasks and achieving comparable performance to single-task models.
5. Extensive experiments on the newly constructed Protein-MT benchmark demonstrate HeMeNet's superior performance over state-of-the-art methods in both single-task and multi-task settings, especially in affinity prediction tasks.
6. The study highlights the potential of multi-task learning in protein structure analysis, suggesting that combining related tasks can significantly enhance the utilization of limited labeled data and improve model robustness.
💻Code: github.com/hanrthu/HeMeNet
📜Paper: ojs.aaai.org/index.php/AAAI/…
#ProteinStructure #MultiTaskLearning #DeepLearning #ComputationalBiology #AIinBiology
2
8
881
15 Dec 2025
Task-Specific Sparse Feature Masks for Molecular Toxicity Prediction with Chemical Language Models
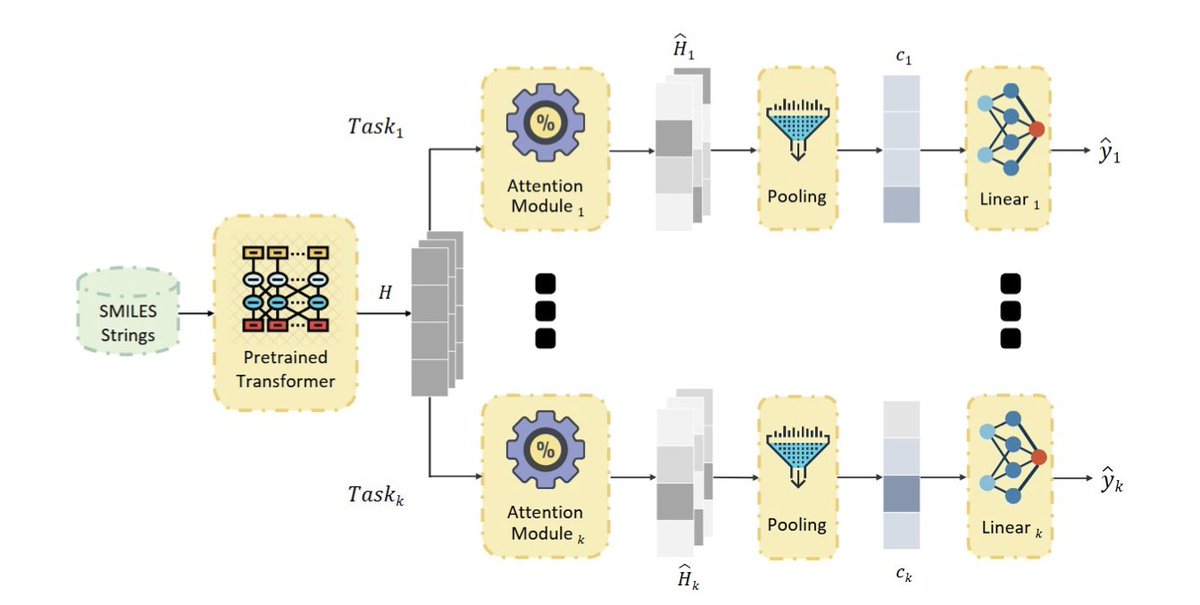
1. A novel multi-task learning (MTL) framework has been proposed to enhance both accuracy and interpretability in molecular toxicity prediction. This approach integrates a shared chemical language model with task-specific attention modules, imposing an L1 sparsity penalty to focus on salient molecular fragments for each toxicity endpoint.
2. The framework outperforms single-task and standard MTL baselines across ClinTox, SIDER, and Tox21 datasets. The sparse attention weights provide intuitive visualizations, revealing specific molecular fragments influencing predictions and enhancing model transparency.
3. The architecture adapts task-specific attention mechanisms into a hard parameter sharing (HPS) framework, allowing each toxicity endpoint to learn tailored representations from a shared chemical embedding. This design mitigates negative transfer and improves stability.
4. Moderate L1 regularization not only improves interpretability but also boosts predictive performance, challenging the traditional trade-off between accuracy and transparency. The model learns generalizable structural patterns rather than memorizing specific scaffolds.
5. The study demonstrates task-specific differentiation and cross-scaffold consistency in attention patterns. For example, the model attends to distinct features within the same molecule for different endpoints and consistently highlights relevant motifs across diverse structures.
📜Paper: arxiv.org/abs/2512.11412v1
#ChemicalLanguageModels #MolecularToxicity #MultiTaskLearning #Interpretability #ComputationalToxicology
1
8
1,132
5 Dec 2025
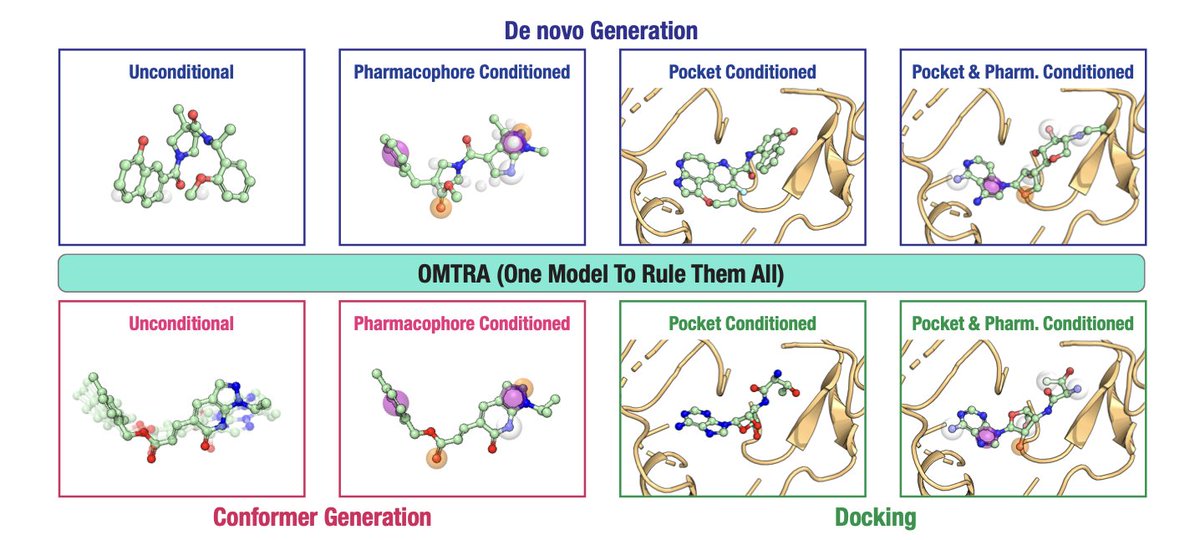
OMTRA: A Multi-Task Generative Model for Structure-Based Drug Design
1. OMTRA introduces a novel multi-task generative model that unifies various tasks in structure-based drug design (SBDD) under one framework. This includes de novo ligand design, docking, and conformer generation, showcasing versatility in handling multiple SBDD-related tasks.
2. The model leverages flow matching, a powerful technique for interpolating between probability distributions, to generate molecular structures. This approach enables the simultaneous modeling of continuous and discrete molecular properties, such as atom positions and types.
3. A key innovation is the ability to condition the model on protein pockets and pharmacophores, allowing for guided design and docking. This feature enhances the accuracy of ligand generation by incorporating prior knowledge of protein-ligand interactions.
4. OMTRA achieves state-of-the-art performance in pocket-conditioned de novo design and docking, outperforming existing models in terms of both chemical plausibility and interaction recovery with proteins.
5. The authors curate a large-scale dataset of 500 million 3D molecular conformers, significantly expanding the chemical diversity available for training. This dataset complements existing protein-ligand data and supports multi-task learning across diverse molecular modalities.
6. Despite the potential benefits of multi-task training, the study finds that its effects are modest and sometimes inconsistent across different tasks. This highlights the ongoing challenge of effectively leveraging transfer learning in molecular generative models.
7. Future work will explore extending OMTRA to generate protein structures, which could support tasks involving flexible or unknown protein conformations. This extension could make OMTRA a more comprehensive tool for realistic drug design scenarios.
💻Code: github.com/gnina/OMTRA
📜Paper: arxiv.org/abs/2512.05080
#DrugDesign #GenerativeModeling #FlowMatching #MultiTaskLearning #StructureBasedDrugDesign #MolecularGeneration
1
4
28
2,193
5 Dec 2025
BioMedGPT-Mol: Multi-task Learning for Molecular Understanding and Generation
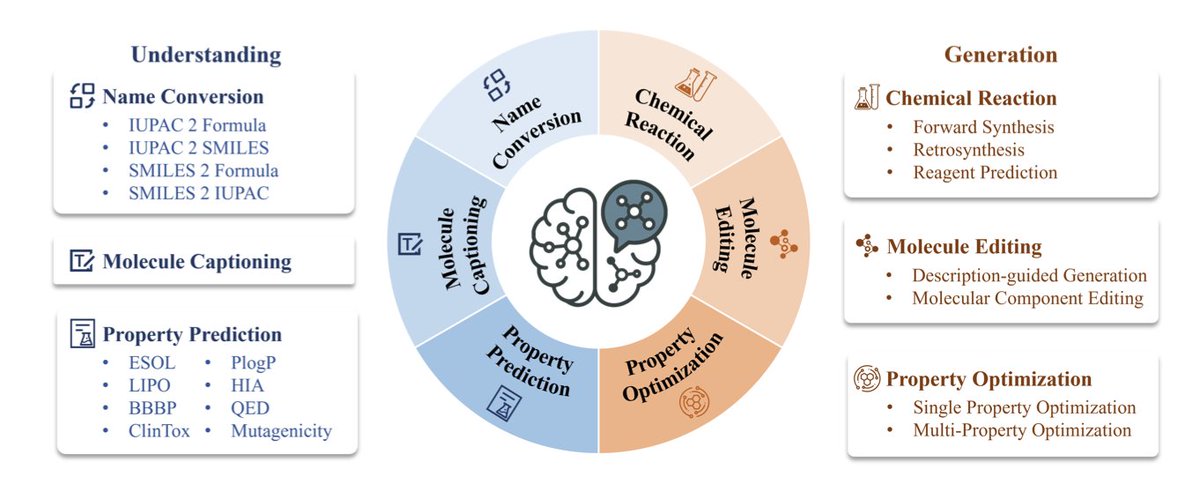
1. This groundbreaking work introduces BioMedGPT-Mol, a specialized molecular language model designed to advance molecular discovery in biomedicine. The model leverages multi-task learning to excel in both understanding and generating molecular structures, setting a new benchmark in the field.
2. The model is trained on a large-scale, comprehensive dataset assembled from various public sources, covering tasks like molecular understanding, chemical reaction prediction, and property optimization. This holistic approach enables BioMedGPT-Mol to capture the intrinsic relationships between molecular components and their properties.
3. A key innovation is the use of special tokens to identify molecular language, enhancing the model's ability to process SMILES strings, IUPAC names, and molecular formulas. This ensures accurate parsing and generation of molecular structures.
4. BioMedGPT-Mol demonstrates remarkable performance on a consolidated benchmark, outperforming both general-purpose LLMs and other chemistry-specific models. It achieves high accuracy in tasks such as name conversion, molecule captioning, and property prediction, showcasing its deep molecular expertise.
5. The model also explores retrosynthetic planning, a challenging task in organic chemistry. Using a three-stage supervised fine-tuning strategy, BioMedGPT-Mol is adapted to function as an end-to-end retrosynthetic planner, achieving competitive results on RetroBench.
6. This work highlights the potential of adapting general-purpose reasoning models for specialized scientific domains. BioMedGPT-Mol serves as a powerful tool for molecular research, with applications ranging from drug discovery to chemical synthesis planning.
📜Paper: arxiv.org/abs/2512.04629v1
#BioMedGPTMol #MolecularLanguageModel #MultiTaskLearning #DrugDiscovery #ChemistryAI
4
5
1,057
22 Nov 2025
Solvaformer: An SE(3)-Equivariant Graph Transformer for Small Molecule Solubility Prediction
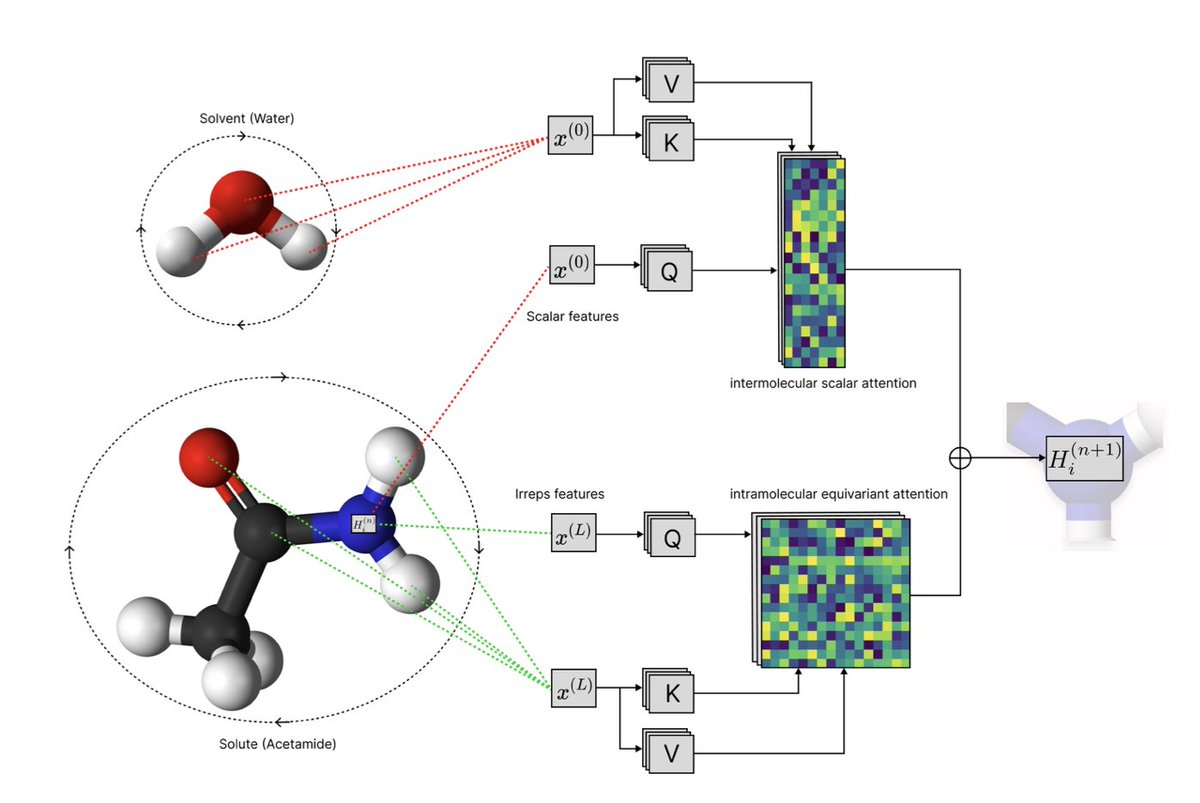
1. Solvaformer is a groundbreaking model that predicts small molecule solubility with high accuracy by integrating geometric awareness and multi-task learning. This approach combines intramolecular SE(3)-equivariant attention and intermolecular scalar attention, enabling the model to capture complex molecular interactions without imposing artificial geometric constraints.
2. The model is trained on both quantum-mechanical data (CombiSolv-QM) and experimental measurements (BigSolDB 2.0) using an alternating-batch regimen. This dual-data strategy allows Solvaformer to leverage the strengths of computational and experimental datasets, enhancing its predictive power and generalizability.
3. Solvaformer outperforms several state-of-the-art models, including EquiformerV2 and various XGBoost-based approaches, achieving an MAE of 0.643 and an RMSE of 0.837 on the BigSolDB 2.0 test set. Its performance is close to the DFT-assisted XGBoost baseline, which is computationally expensive and impractical for large-scale screening.
4. A key innovation of Solvaformer is its interpretability. Case studies demonstrate that the model can distinguish between intramolecular and intermolecular hydrogen bonding patterns in positional isomers, such as salicylic acid and 4-hydroxybenzoic acid. This ability to capture chemically coherent interactions makes Solvaformer a powerful tool for understanding solubility differences.
5. Solvaformer’s architecture and training strategy offer a practical balance of accuracy, scalability, and interpretability, making it suitable for high-throughput screening and hypothesis-driven analysis in solution-phase modeling. Future work includes enhancing the dataset with more geometric isomers and exploring multiple conformers to improve geometric sensitivity.
📜Paper: arxiv.org/abs/2511.09774
#Solvaformer #SE3Equivariant #SolubilityPrediction #MultiTaskLearning #ComputationalChemistry #Interpretability
2
17
56
4,414
16 Oct 2025
Multitask finetuning and acceleration of chemical pretrained models for small molecule drug property prediction
1. A new study explores the application of multitask learning in finetuning chemical pretrained models, specifically focusing on small molecule drug property prediction. The research shows significant performance improvements over non-pretrained models, especially in large data scenarios.
2. The study introduces KERMT, an enhanced version of the GROVER model, which demonstrates superior performance in multitask finetuning. KERMT outperforms other models more significantly as the data size increases, contrary to previous hypotheses.
3. The researchers published two multitask ADMET data splits to enable more accurate benchmarking of multitask deep learning methods for drug property prediction. This provides a valuable resource for future research and development in this area.
4. An accelerated implementation of the KERMT model is provided, allowing for large-scale pretraining, finetuning, and inference in industrial drug discovery. This advancement could significantly speed up the drug discovery process.
5. The study highlights the importance of preserving graphical structure in molecular representations for predicting molecular properties. Pretraining on molecular graphs yields more expressive representations compared to basic SMILES strings.
6. The research also investigates the scaling behavior of KERMT, showing that it performs better at larger full data sizes. This suggests that KERMT benefits from larger parameter spaces and more data, which is crucial for its effectiveness in drug discovery applications.
7. The study explores the generalization capabilities of KERMT across different chemical spaces, finding that it consistently outperforms Chemprop across all similarity regimes. This indicates KERMT's ability to generalize to less similar chemical spaces.
8. The authors provide insights into the pretraining tasks and their impact on downstream performance. They suggest that further research is needed to identify optimal pretraining tasks and data curation strategies for improved performance.
9. The study concludes with recommendations for using KERMT multitask on medium to large data sizes and KPGT single-task on small data sizes for ADMET property prediction, based on empirical results.
📜Paper: arxiv.org/abs/2510.12719v1
💻Code: github.com/NVIDIA-Digital-Bi…
#MultitaskLearning #DrugDiscovery #ChemicalPretraining #DeepLearning #ADMETPrediction

1
7
941
16 Oct 2025
Structured Chemical Reaction Modeling with Multitask Graph Neural Networks
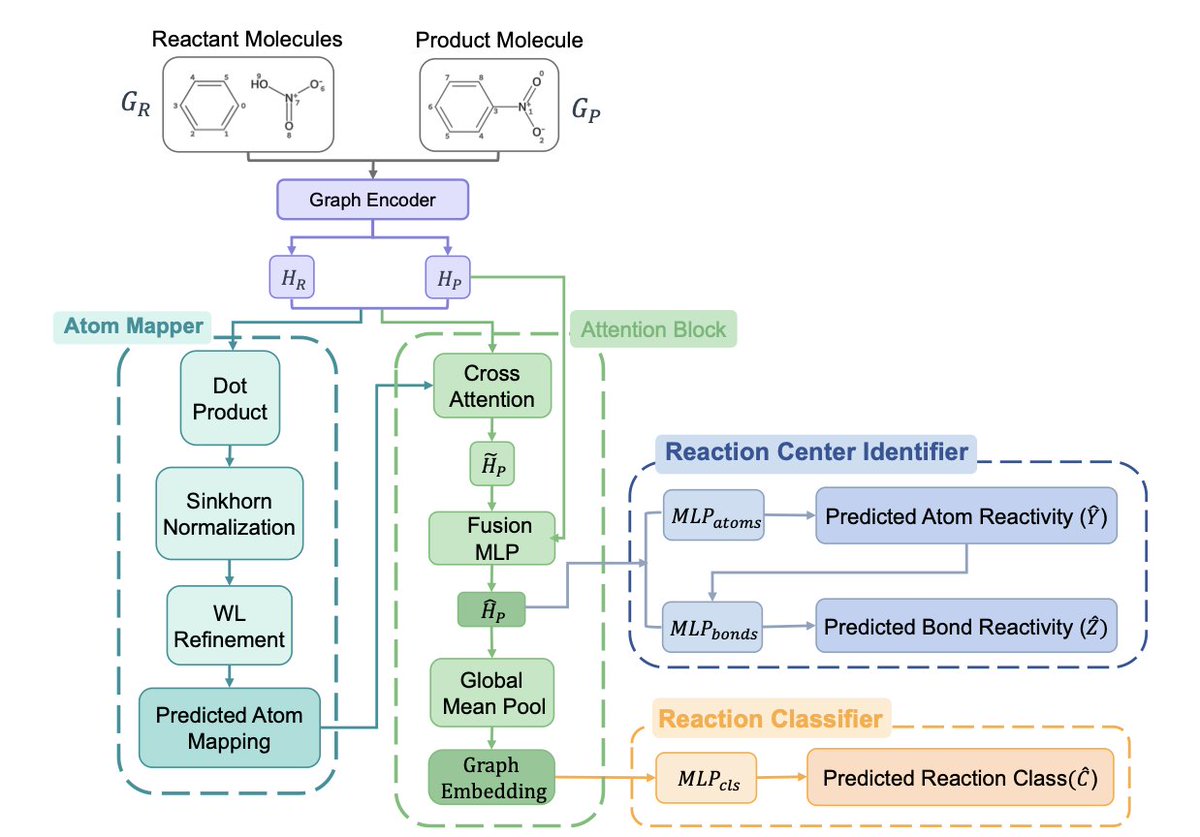
1. A new study proposes MARCC, a multitask graph neural network framework that integrates atom mapping, reaction center prediction, and reaction classification into a single model. This approach leverages the interdependencies between these tasks to improve accuracy and interpretability in chemical reaction modeling.
2. MARCC uses a shared graph neural network encoder for reactant and product molecules, ensuring consistent embeddings across tasks. It employs a differentiable atom-mapping module to align product atoms with their reactant counterparts, guiding cross-attention mechanisms that enrich product embeddings with mapped reactant context.
3. The study demonstrates that multitask learning significantly enhances performance. MARCC achieves state-of-the-art results on the USPTO-50K benchmark, with near-perfect reaction center prediction accuracy (99.1%) and high classification accuracy (97.2%). The full multitask setup consistently outperforms single-task and dual-task configurations.
4. MARCC’s atom mapping accuracy (98.2%) is on par with specialized systems, while also excelling in center prediction and classification. This highlights the strength of multitask learning, where all three tasks are interdependent and jointly improve overall model performance.
5. The authors argue that multitask learning not only improves accuracy but also enhances interpretability by producing a unified structural narrative of chemical reactions. This structured output aligns with how chemists reason about reactions, offering valuable insights for reaction understanding and template discovery.
📜Paper: biorxiv.org/content/10.1101/…
#MultitaskLearning #GraphNeuralNetworks #ChemicalReactions #ReactionModeling #MachineLearning #Biochemistry #MolecularScience
2
13
963
16 Oct 2025
M 3ST-DTI: A Multi-Task Learning Model for Drug-Target Interactions Based on Multi-Modal Features and Multi-Stage Alignment
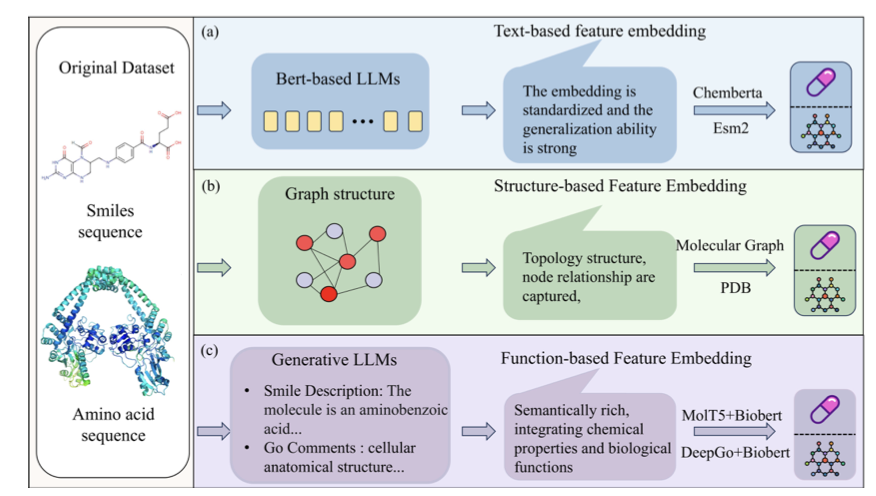
1. A novel multi-task learning model called M 3ST-DTI has been proposed for predicting drug-target interactions (DTI) with enhanced accuracy and generalization. This model stands out by integrating multi-modal features and employing a multi-stage alignment strategy, significantly improving the predictive performance compared to existing methods.
2. M 3ST-DTI incorporates three types of features - textual, structural, and functional - to capture comprehensive information for DTI prediction. It uses self-attention mechanisms and a hybrid pooling graph attention module to enhance intra-modal representations, which is a crucial step for extracting detailed and meaningful features from each modality.
3. The model features an early fusion stage that combines multi-modal features with Gram loss as a structural constraint for effective alignment. This approach ensures that the features from different modalities are semantically aligned in the embedding space, laying a solid foundation for subsequent interactions and predictions.
4. In the late fusion stage, M 3ST-DTI employs a BCA module to capture fine-grained interactions within each modality and a deep orthogonal fusion module to reduce feature redundancy. This combination allows the model to refine the integration of cross-modal representations and enhance the discriminability of the final feature representation.
5. Extensive evaluations on the BindingDB dataset demonstrate that M 3ST-DTI consistently outperforms state-of-the-art methods across various metrics, including accuracy, F1-score, AUROC, and AUPRC. The results highlight the robustness and effectiveness of the multi-modal, multi-stage fusion framework in DTI prediction.
6. UMAP visualizations reveal the progressive optimization of feature representations throughout the fusion stages. From raw single-modal inputs to the final joint representations, the model shows increasingly distinct boundaries between positive and negative samples, significantly enhancing class separability.
📜Paper: arxiv.org/abs/2510.12445
#DrugTargetInteraction #MultiTaskLearning #MultiModalFeatures #DeepLearning #Bioinformatics
1
5
806
10 Oct 2025
ATOM: A Pretrained Neural Operator for Multitask
Molecular Dynamics
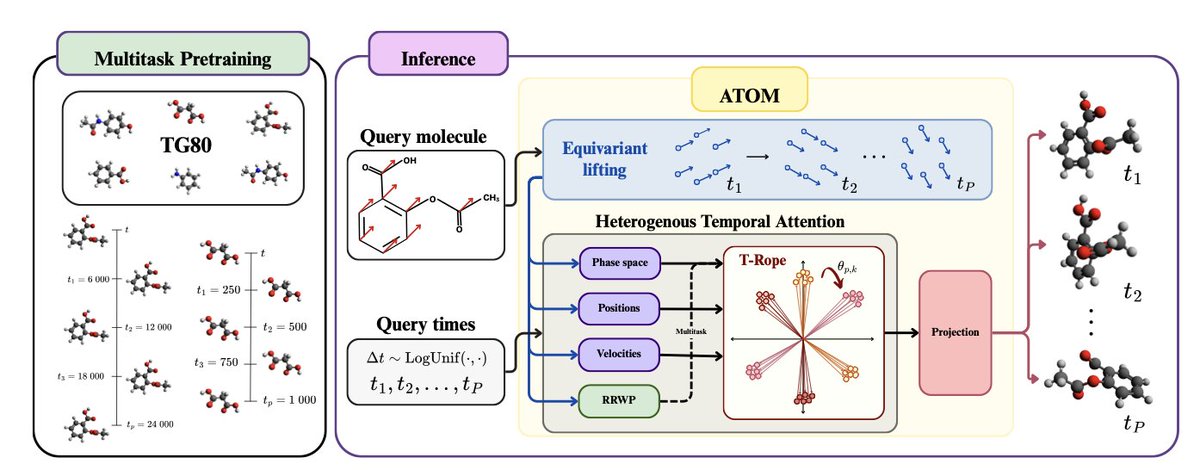
1. ATOM is a novel pretrained neural operator designed for multitask molecular dynamics simulations, addressing limitations of existing methods that enforce strict equivariance and rely on sequential rollouts, which restrict flexibility and efficiency.
2. The model introduces a quasi-equivariant design that requires no explicit molecular graph, allowing for more flexible and efficient simulations. It employs a temporal attention mechanism for accurate parallel decoding of multiple future states.
3. ATOM achieves state-of-the-art performance on established single-task benchmarks like MD17, RMD17, and MD22. After multitask pretraining on the new TG80 dataset, it shows exceptional zero-shot generalization to unseen molecules across varying time horizons.
4. The TG80 dataset is a large, diverse, and numerically stable MD dataset with over 2.5 million femtoseconds of trajectories across 80 compounds, curated to support operator pretraining across chemicals and timescales.
5. ATOM’s design innovations include an equivariant lifting layer for symmetry-aware features, heterogeneous temporal attention, and a temporal rotary position embedding (T-RoPE) for robust temporal predictions.
6. The model demonstrates significant improvements in both single-task and multitask settings, with an average improvement of 39.75% in multitask performance on unseen molecules and timeframes.
7. ATOM represents a significant step toward accurate, efficient, and transferable molecular dynamics, showcasing the potential of quasi-equivariance designs and zero-shot generalization in this field.
📜Paper: arxiv.org/abs/2510.05482
#MolecularDynamics #NeuralOperators #Pretraining #MultitaskLearning #ComputationalBiology
2
4
987

9 Oct 2025
⚡ Smarter AI for Cancer Care: Transformers in Radiotherapy Planning
Radiation therapy saves lives, but planning the right dose distribution is one of the most challenging tasks for radiologists. Traditionally, plans are adjusted iteratively — slow, subjective, and prone to variation.
This 2023 study introduces a Transformer-Embedded Multi-Task Dose Prediction (TransMTDP) model, designed to make radiotherapy planning more accurate, efficient, and consistent.
🔬 Key innovations:
• Multi-task learning: Combines 3 tasks — fine-grained dose prediction, isodose lines prediction, & gradient map prediction.
• Transformer embedding: Captures long-range dependencies in dose maps, reflecting anatomical symmetries & radiation gradients.
• Consistency constraints: Improves alignment between auxiliary tasks and main dose prediction.
📊 Results:
• Outperformed state-of-the-art models on both in-house rectum cancer data & public head-and-neck datasets.
• Provided stable, fine-grained dose maps with better clinical reliability.
• Reduced subjectivity & workload for radiologists, accelerating treatment planning.
💡 Why it matters:
TransMTDP supports radiologists with AI-powered predictions, ensuring more personalized, precise, and timely cancer treatment, especially for complex cases like head & neck cancers.
Who It’s For
• Radiation oncologists & radiologists
• AI in healthcare researchers
• Oncology clinicians & treatment planners
• Medical imaging professionals
• Cancer care foundations & policy makers
• AI/ML engineers working in medical imaging
📘 Read the article for free now:
👉 worldscientific.com/doi/10.1…
💸 Recommend the journal to your library today!
#CancerCare #Radiotherapy #AIHealthcare #Transformers #DeepLearning #DosePrediction #Oncology #Radiology #MedicalAI #CancerTreatment #MultiTaskLearning #HealthcareInnovation #AIinMedicine #PrecisionOncology #MachineLearning #HealthTech #OncoAI #ArtificialIntelligence #MedicalImaging #OncologyResearch
@NatureMedicine @TheLancetOncol @JAMA_current @MIT_CSAIL @deepmind @GoogleHealth @NVIDIAAI @OpenAI @RSNA @AIMedicalGroup @Radiology_News @OncoAlert @MDAndersonNews @cancerresearchuk @ASCO @MITechReview @ai_in_healthcare @neuroai_news @oncologynewsint @HealthTechMag

2
172
6 Oct 2025
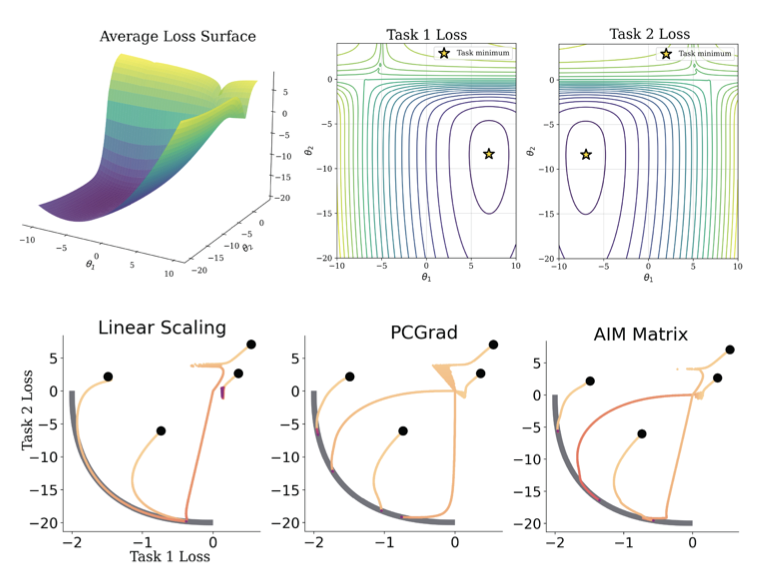
AIM: Adaptive Intervention for Deep Multi-task Learning of Molecular Properties
1. A new optimization framework called AIM is proposed to tackle the challenge of optimizing multiple conflicting molecular properties simultaneously, a common bottleneck in drug discovery. AIM learns a dynamic policy to mediate gradient conflicts in multi-task learning, which is particularly effective in data-scarce regimes.
2. AIM introduces a novel augmented objective composed of dense, differentiable regularizers. This objective guides the policy to produce updates that are geometrically stable and dynamically efficient, prioritizing progress on the most challenging tasks. The policy is trained jointly with the main network.
3. The policy in AIM has two variants: scalar policy and matrix policy. The matrix policy learns a unique threshold for each pair of tasks, allowing the model to capture nuanced pairwise relationships and providing an interpretable diagnostic tool to analyze inter-task relationships.
4. Experiments on the QM9 benchmark and a complex targeted protein degrader (TPD) ADME benchmark demonstrate AIM's effectiveness. AIM achieves statistically significant improvements over multi-task baselines, especially in low-data regimes. The learned policy matrix offers insights into the MTL process.
5. In the QM9 benchmark, AIM significantly outperforms all baselines on both Mean Rank and ∆m% in the 10k subset. On the TPD ADME benchmark, AIM delivers state-of-the-art performance, particularly in the more data-scarce regime, showing its strong ability to find a robust and well-balanced solution across competing tasks.
6. AIM's adaptive policy is most valuable when data is limited. As the amount of training data increases, simpler heuristics become more competitive. AIM's interpretable policy serves as a diagnostic tool that offers insights into the relationships between tasks, building confidence in the model and its optimization strategies.
📜Paper: arxiv.org/abs/2509.25955
#AIM #MultiTaskLearning #MolecularProperties #DrugDiscovery #DeepLearning #Optimization
4
12
1,021
3 Oct 2025
Enhancing Molecular Dipole Moment Prediction with Multitask Machine Learning
1. Researchers have developed a multitask machine learning strategy to improve the prediction of molecular dipole moments by incorporating Mulliken atomic charges as an auxiliary task. This approach enhances model accuracy by up to 30%, despite Mulliken charges being less accurate but providing valuable qualitative information about charge distribution.
2. The study highlights the effectiveness of multitask learning in quantum chemistry applications. By using a small weight for atomic charges in the loss function, the model learns a more physically grounded representation of charge distributions, leading to improved accuracy and consistency in dipole moment predictions.
3. The method uses the QM9 and QMugs datasets to benchmark performance. The results show significant improvements in dipole prediction accuracy when atomic charges are included in the training process, even when using average charge values, demonstrating the robustness of the multitask learning approach.
4. The Hierarchical Interacting Particle Neural Network (HIP-NN) is employed to capture both local and global interactions within molecules. This architecture allows the model to integrate short-range and long-range interactions, resulting in more accurate predictions of global molecular properties such as dipole magnitudes.
5. The findings suggest that even computationally inexpensive and less accurate auxiliary data can provide valuable qualitative insights for machine learning models, ultimately enhancing their predictive power for molecular properties. This approach holds promise for advancing data-driven molecular modeling in computational chemistry and materials science.
📜Paper: arxiv.org/abs/2509.22435v1
#MachineLearning #QuantumChemistry #MolecularModeling #MultitaskLearning
3
690

18 Sep 2025
Equation: Tasks×SharedReps → generalization↑ 🧠
Cut compute; share features; curb overfit; tune loss weights; separate heads; audit conflicts 🛠️ - GLCND.IO
Explore → glcnd.io/unlocking-multi-tas…
#AI #MultiTaskLearning #DeepLearning
2
17
8 Sep 2025
Quantum-Enhanced Multi-Task Learning with Learnable Weighting for Pharmacokinetic and Toxicity Prediction
1. A novel framework, QW-MTL, has been proposed to enhance ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction in drug discovery. This is achieved through a combination of quantum-informed molecular representations and a learnable task weighting mechanism, which significantly improves predictive performance across multiple tasks.
2. QW-MTL integrates quantum chemical descriptors with traditional molecular descriptors to enrich the input features for machine learning models. This hybrid approach provides a more comprehensive representation of molecular properties, leading to better predictions of pharmacokinetic and toxicity profiles.
3. The framework introduces a dynamic task weighting strategy based on sample scale. This allows the model to adaptively balance the contribution of each task during training, addressing the issue of task imbalance and improving overall performance in multi-task learning scenarios.
4. QW-MTL has been evaluated on 13 benchmark tasks from the Therapeutics Data Commons (TDC) and has shown superior performance compared to single-task learning baselines. It achieves state-of-the-art results on multiple tasks, demonstrating the effectiveness of the proposed approach.
5. The model maintains a comparable parameter count to baseline models while significantly improving inference efficiency. This makes QW-MTL a scalable and efficient solution for large-scale molecular screening and property evaluation in drug discovery.
📜Paper: arxiv.org/abs/2509.04601
#QuantumEnhancedLearning #MultiTaskLearning #ADMETPrediction #DrugDiscovery #MachineLearning

3
1,218

8 Sep 2025
🔥 Read our Paper
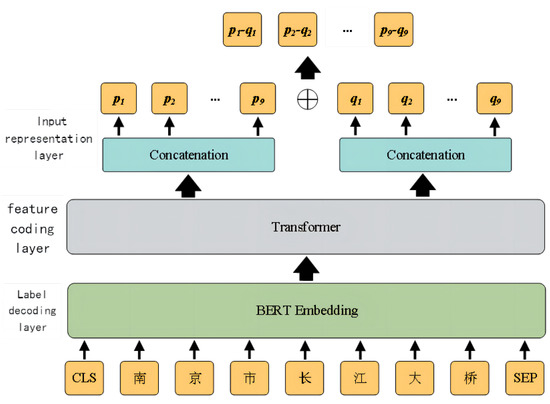
📚 Chinese Named Entity Recognition Model Based on Multi-Task Learning
🔗 mdpi.com/2076-3417/13/8/4770
👨🔬 by Qin Fang et al.
#multitasklearning #Chinesenamedentityrecognition
1
2
101