
Joined July 2017
- Tweets 318
- Following 952
- Followers 146
- Likes 1,235
19 Photos and videos









Yiannis Galdadas retweeted

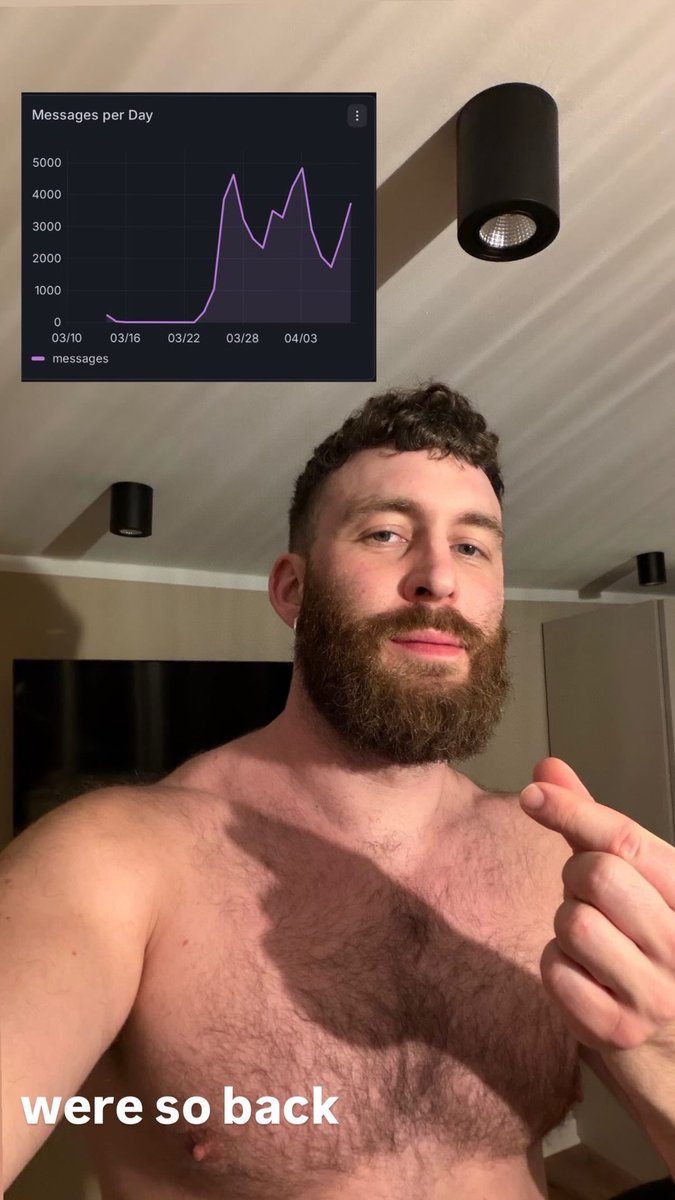

I’m so happy messaging is fixed on meetmarket! You‘ll now find messages and requests that were undelivered due to a connection issue. You can also now tap a message request to expand the profile before accepting. Check it out and let me know how it goes 🥩✨

1
1
86
36,343
Yiannis Galdadas retweeted

Our heavily updated final version of ROCKET for experiment-guided structure modeling is out in @naturemethods!
New frontiers like high-throughput fragment screening and mid-res tomography are forcing a rethink of how experiments are modeled. ROCKET OpenFold proposes a solution👇
Apr 1

ROCKET makes AlphaFold context-aware. We iteratively steer prediction to agree with experiment (cryo-EM, crystallography) at inference time, no retraining. Structure determination becomes a search where ML priors and experiment productively combine and inform each other. 2/15
1
41
212
18,358
Yiannis Galdadas retweeted

17 Dec 2025
PXDesign is now Open-Source (Apache 2.0)!
SOTA hit rates. Efficient binder generation.
🔹 Updated Wet-lab data: up to 82% SR
🔹 Positive feedback from Web Server users
🔗 Code: github.com/bytedance/PXDesig…
🔗 Server: protenix-server.com
#ProteinDesign #OpenSource #GenerativeAI

2
31
118
29,169
Yiannis Galdadas retweeted

14 Dec 2025
Looks excellent! Fast and accurate Boltzmann Generators by @danyalrehman17 @tara_aksa @ag27182 @AlexanderTong7 @Yoshua_Bengio @Mila_Quebec
12 Dec 2025

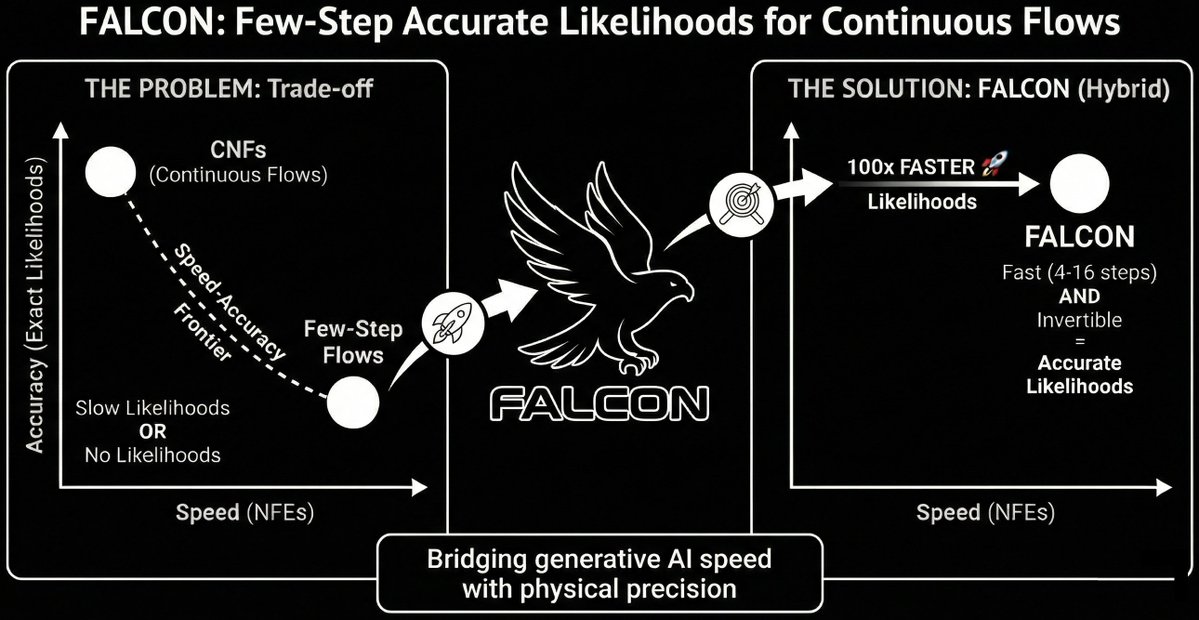
🧵1/9 Flow maps have sped up sampling 100x, but can we speed up likelihoods too? 🚀
For scientific applications like Boltzmann Generators, fast samples aren't enough—you need exact likelihoods. That has been the major bottleneck.
Introducing FALCON 🦅: A solution for fast AND accurate likelihoods. 🧵👇
🔗: arxiv.org/pdf/2512.09914
👥: @tara_aksa @ag27182 @AlexanderTong7 @Yoshua_Bengio @Mila_Quebec
1
13
87
9,976
Yiannis Galdadas retweeted

3 Dec 2025
Our December issue is live!
nature.com/nchembio/volumes/…
The cover depicts a diffusion-based generative deep learning pipeline for de novo design of a macrocyclic peptide targeting the protein, myeloid cell leukemia 1

1
14
88
6,647
Yiannis Galdadas retweeted

3 Dec 2025
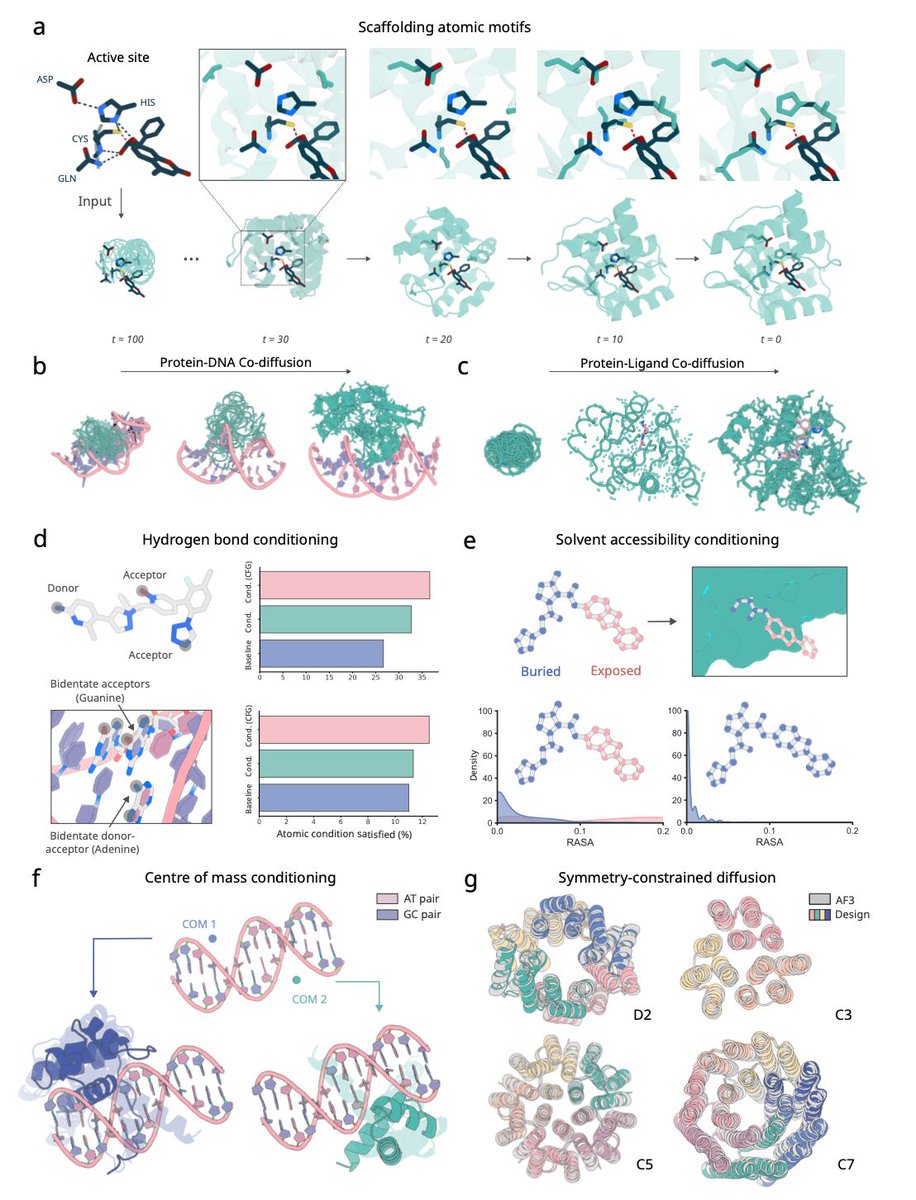
RFdiffusion3 now available! De novo protein design against any molecule
Try it on @tamarindbio today
RF3 shows success in designing de novo proteins against all-atom targets, including proteins, DNA and small molecules with diverse applications.
5
60
268
15,102
Yiannis Galdadas retweeted

24 Nov 2025
incredibly detailed technical blog just dropped on the anatomy of BoltzGen 🧬
made for ML people, but covering everything from molecular representations to diffusion-based generation of protein binders
crazy good interactive visuals
👏👏 @ludocomito

6
36
231
21,943
Yiannis Galdadas retweeted
10 Nov 2025
Fantastic work by Michael Plainer and friends. Energy-based diffusion models to ensure that the denoising distribution equals exp(-u(x)), with the energy u(x). A keystone for connecting molecular dynamics, statistical mechanics and generative AI.
10 Nov 2025

(1/n) Can diffusion models simulate molecular dynamics instead of generating independent samples?
In our NeurIPS2025 paper, we train energy-based diffusion models that can do both:
- Generate independent samples
- Learn the underlying potential 𝑼
🧵👇
arxiv.org/abs/2506.17139
1
60
397
35,476
Yiannis Galdadas retweeted

3 Nov 2025
Introducing Molview - the ipython/jupyter widget version of nano-protein-viewer🔍:
12
167
1,076
38,616
Yiannis Galdadas retweeted
26 Oct 2025
BoltzGen: New generalized binder design protocol, with wet lab validation on diverse targets! (From Boltz team collaborators)
On @tamarindbio right now!
The authors test only 15 nanobody designs against each of 9 targets. These targets are selected for their high dissimilarity from any protein with an existing bound structure. With 6 of the 9 finding nanomolar binders. This 67% success rate holds for protein designs.

1
49
256
13,828
Yiannis Galdadas retweeted

26 Oct 2025
Thrilled to finally see BoltzGen, our new state-of-the-art all-atom binder design model, coming out fully open-source after a very extensive experimental validation with many top academic and industry labs! 🧬
The diversity of the experiments is unprecedented, spanning binder modalities from nanobodies to disulfide-bonded peptides and including targets ranging from disordered proteins to small molecules. These experiments demonstrate state-of-the-art performance, for example, a 67% success rate at designing nanomolar nanobody binders against several novel targets with only 15 or fewer designs. 🚀
Incredible work from an amazing team led by @HannesStaerk! 🤗
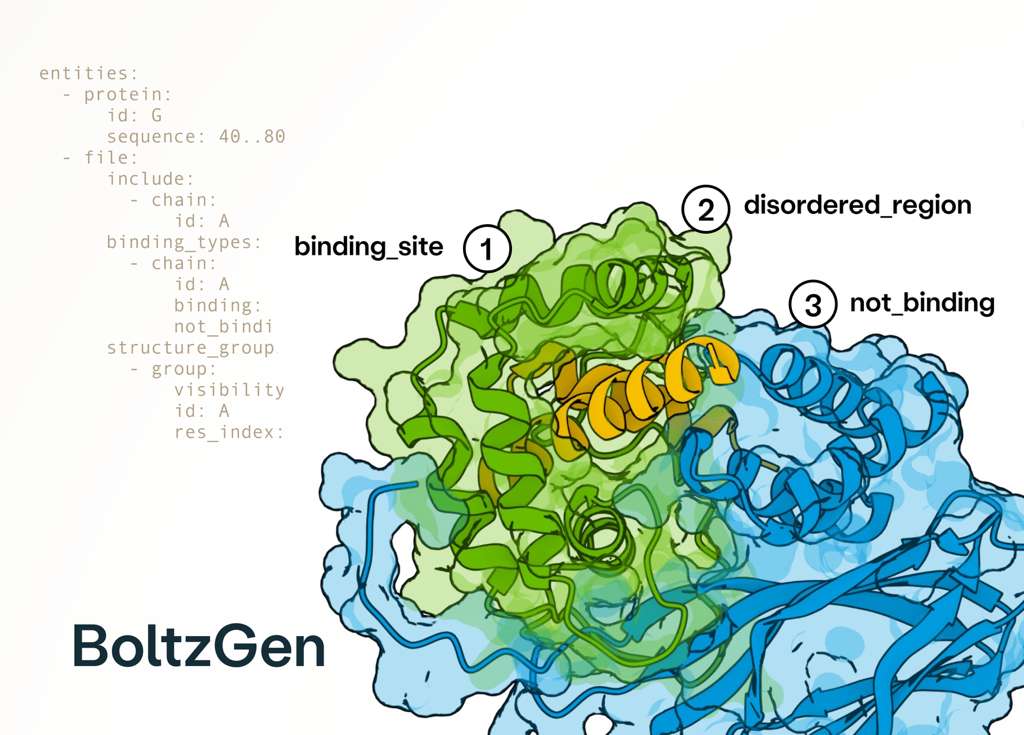
4
79
355
42,027
Yiannis Galdadas retweeted

17 Oct 2025
Researchers have discovered a mechanism that keeps the physiologically important Wnt signaling pathway from becoming overly active, which can occur during the initiation and metastasis of various types of #cancer. @SciSignal scim.ag/4n84XV6
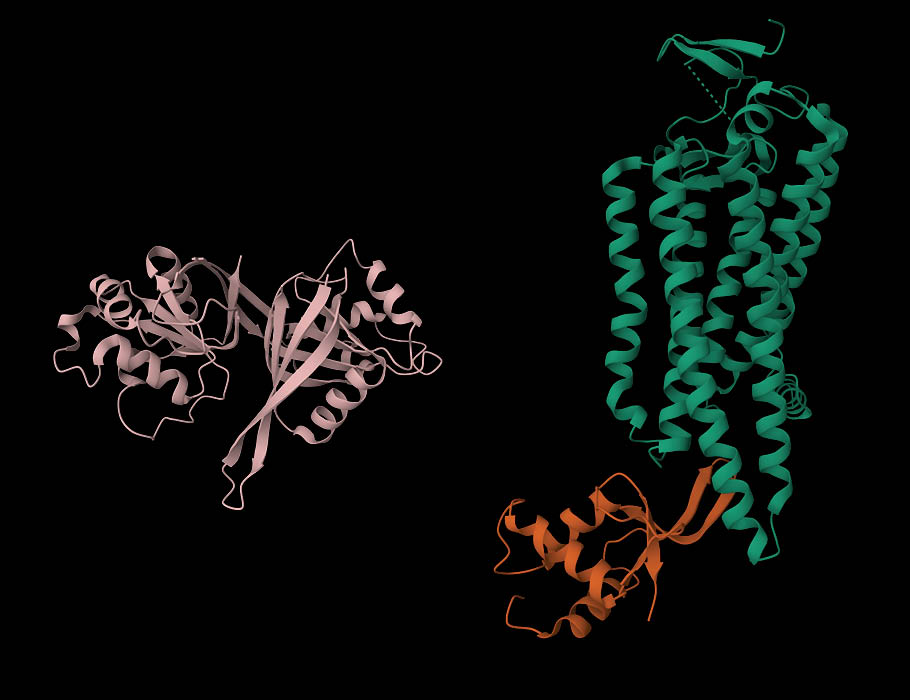
6
38
194
26,088
Yiannis Galdadas retweeted

28 Sep 2025
A Foundation Chemical Language Model for Comprehensive Fragment-Based Drug Discovery
1. FragAtlas-62M is a groundbreaking chemical language model specifically designed for fragment-based drug discovery. It is trained on the largest fragment dataset to date, comprising over 62 million molecules from the ZINC-22 database. This model achieves an impressive 99.90% chemical validity in generated fragments, making it a powerful tool for medicinal chemistry.
2. The model not only maintains a high coverage of known ZINC fragments (53.55%) but also generates 22.04% novel structures with practical relevance. This balance between rediscovery and novelty is crucial for fragment-based drug discovery, as it ensures both the reliability of known fragments and the potential for new discoveries.
3. FragAtlas-62M is built on a GPT-2 architecture with 42.7M parameters. It uses a 128-token context window and is trained using HuggingFace Transformers. The model's architecture and training methodology are optimized for fragment-level SMILES modeling, ensuring efficient and high-throughput generation capabilities.
4. The model's performance is validated across 12 molecular descriptors and three fingerprint methods. The generated fragments closely match the training distribution, with all effect sizes being less than 0.4. This indicates that the model generalizes well and maintains the key properties of the training set without systematic bias.
5. FragAtlas-62M demonstrates substantial overlap in chemotype space between novel and rediscovered molecules, as shown by t-SNE visualizations and distance analysis. The distance ratios (NR/NN and NR/RR) are consistently near 1.0 across all three fingerprint types, indicating minimal distributional shifts.
6. The model is released with training code, preprocessed data, documentation, and model weights, making it accessible for further research and practical applications. This open release lowers the barrier to entry for groups with modest computational resources and encourages rapid follow-up work and experimental validation.
7. While FragAtlas-62M is a significant advancement, it has limitations. It does not explicitly model stereochemistry, geometric relationships, or fragment-to-fragment connectivity rules. Future work should focus on integrating conditional controls, structural information, and methods for automated molecule construction to broaden its practical applications.
📜Paper: arxiv.org/abs/2509.19586
#FragAtlas62M #ChemicalLanguageModel #FragmentBasedDrugDiscovery #MedicinalChemistry #AIinDrugDiscovery #OpenSource #Research
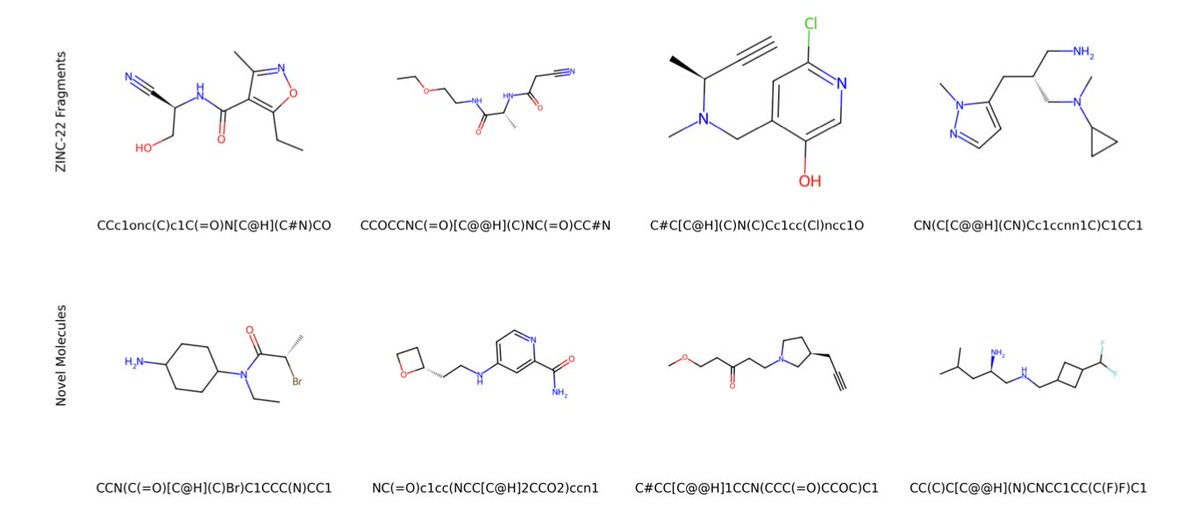
1
8
26
2,839
Yiannis Galdadas retweeted

10 Sep 2025
Really excited to release nvMolKit! GPU drop-in replacements for RDKit functions, free and open source!
9 Sep 2025

🔥nvMolKit landed today🔥
Morgan Fingerprinting, Tanimoto/Cosine similarity and MMFF geometry optimization and conformer generation on GPU, 10-3000x faster. Screen millions of SMILES before coffee & upsize your QSAR pipelines.
🚀 Which dataset operation will you accelerate first?
#GPU #cheminformatics #drugdiscovery
1
10
61
6,101
Yiannis Galdadas retweeted

10 Sep 2025
µProtein framework: Deep learning RL for smarter protein engineering
Engineering proteins with new or improved functions could transform medicine, agriculture, and biotechnology. The challenge is that the space of possible mutations is astronomically large, and experiments can only probe a tiny fraction of it. While high-throughput methods like deep mutational scanning help, they mainly cover single mutations, leaving the vast landscape of combined mutations unexplored. This is where computational methods and AI step in.
In a new work, Haoran Sun and coauthors introduce µProtein, a framework that combines two components: µFormer, a deep learning model that predicts how mutations affect protein function, and µSearch, a reinforcement learning algorithm that uses µFormer’s predictions to navigate the mutational landscape efficiently. Together, they enable the discovery of multi-mutation protein variants with improved properties, starting only from single-mutation data. The team applied µProtein to optimize the enzyme TEM-1 β-lactamase against the antibiotic cefotaxime, identifying novel multi-mutant variants with activities surpassing all previously known ones.
This work demonstrates how combining machine learning with experimental biology can accelerate protein engineering. By efficiently searching through enormous mutational spaces, µProtein opens new opportunities for enzyme optimization, antibody design, and anticipating drug-resistance mutations. It provides a strong step toward a future where AI not only interprets protein data but also guides the creation of new biological functions.
Paper: nature.com/articles/s42256-0…

1
34
267
15,580
Yiannis Galdadas retweeted

29 Aug 2025
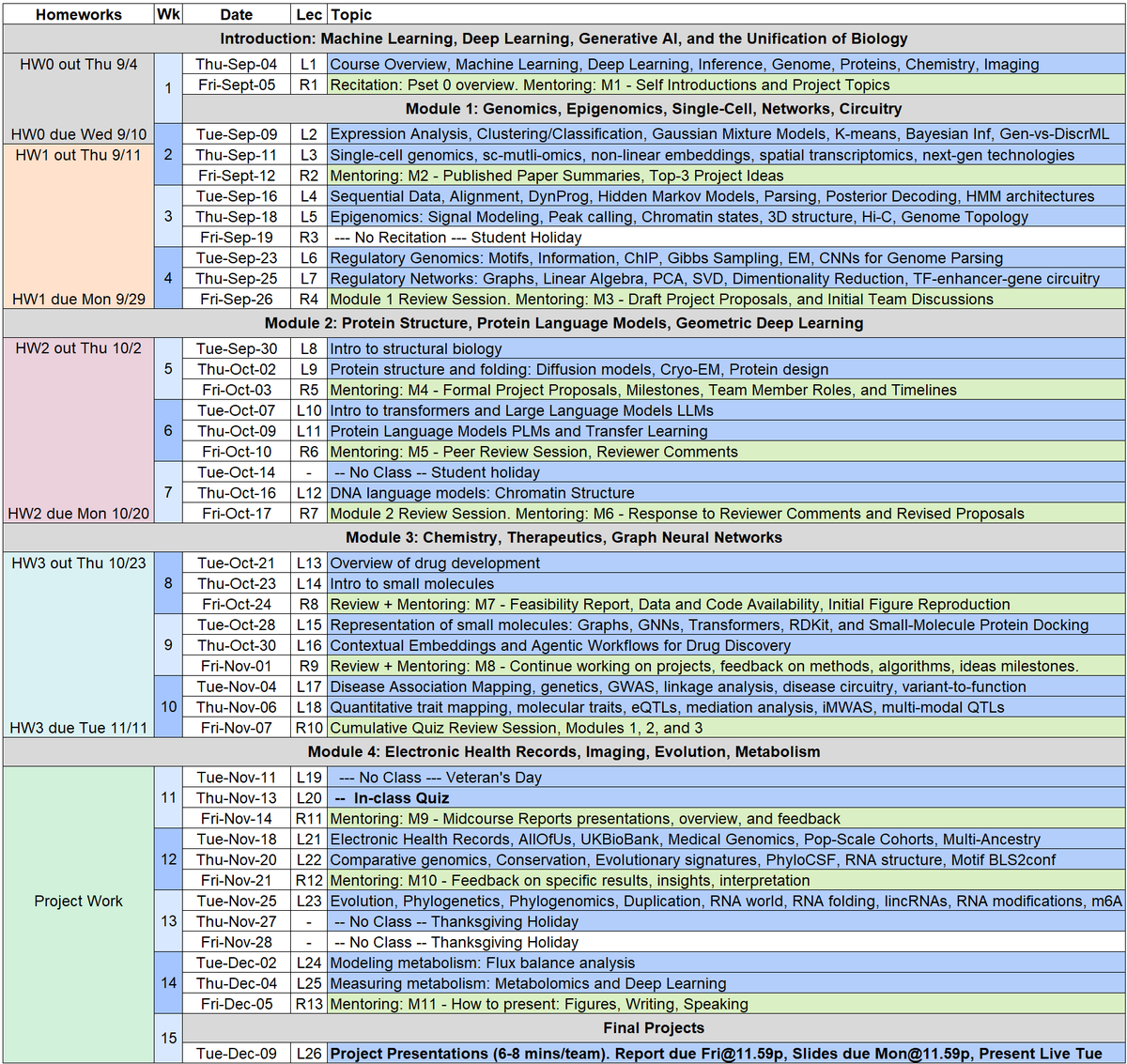
MIT Course announcement: Machine Learning for Computational Biology #MLCB25
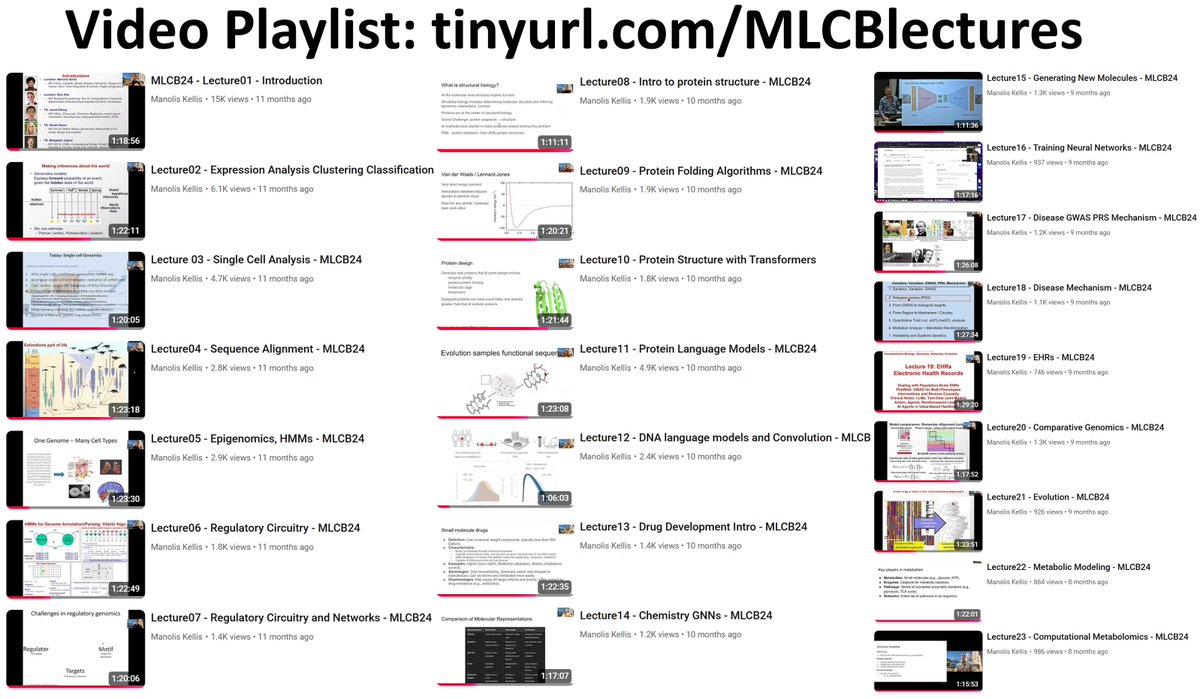
Fall'24 Lecture Videos: tinyurl.com/MLCBlectures
Fall'24 Lecture Notes: tinyurl.com/MLCB24notes
(a) Genomes: Statistical genomics, gene regulation, genome language models, chromatin structure, 3D genome topology, epigenomics, regulatory networks.
(b) Proteins: Protein language models, structure and folding, protein design, cryo-EM, AlphaFold2, transformers, multimodal joint representation learning.
(c) Therapeutics: Chemical landscapes, small-molecule representation, docking, structure-function embeddings, agentic drug discovery, disease circuitry, and target identification.
(d) Patients: Electronic health records, medical genomics, genetic variation, comparative genomics, evolutionary evidence, patient latent representation, AI-driven systems biology.
Foundations and frontiers of computational biology, combining theory with practice. Generative AI, foundation models, machine learning, algorithm design, influential problems and techniques, analysis of large-scale biological datasets, applications to human disease and drug discovery.
First Lecture: Thu Sept 4 at 1pm in 32-144
With: Prof. Manolis Kellis @manoliskellis, Prof. Eric Alm @ejalm, TAs: Ananth Shyamal, Shitong Luo @luost26
Course website: compbio.mit.edu/mlcb
@MIT @MITEECS @MITdeptofBE @MITCSBPhD @MIT_CSAIL @Harvard @HarvardMed @BroadInstitute


6 Dec 2018

Today was my last lecture for @MIT #ComputationalBiology: #Genomes, #Networks, #Evolution, #Health. I recorded each and immediately posted online here: youtube.com/playlist?list=PL…
Please do share, and let me know which topics need more explanations, clarifications, and corrections!
21
470
2,204
253,113
Yiannis Galdadas retweeted

25 Aug 2025
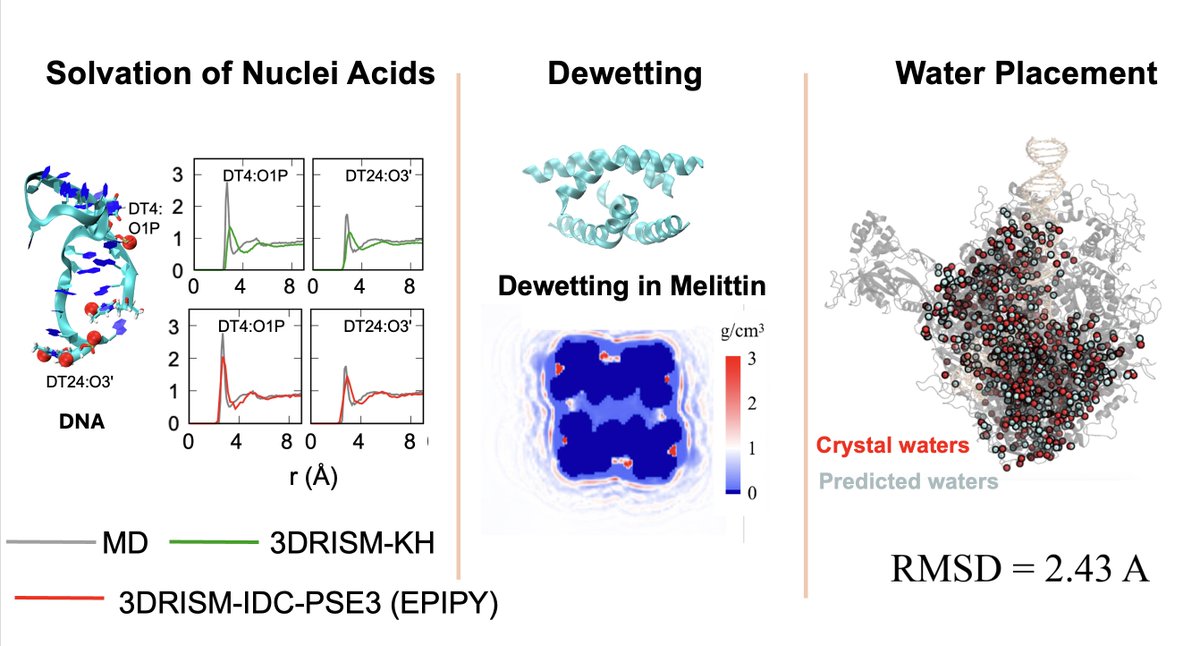
Check out our new Python package, EPIPY!
It streamlines 3DRISM calculations—great for studying RNA/DNA solvation, predicting dewetting, and placing water molecules, etc.
Github: github.com/EPISOLrelease/EPI…
Preprint: chemrxiv.org/engage/chemrxiv…
Congs to @p_swanson123 @SiqinCao!
25 Aug 2025

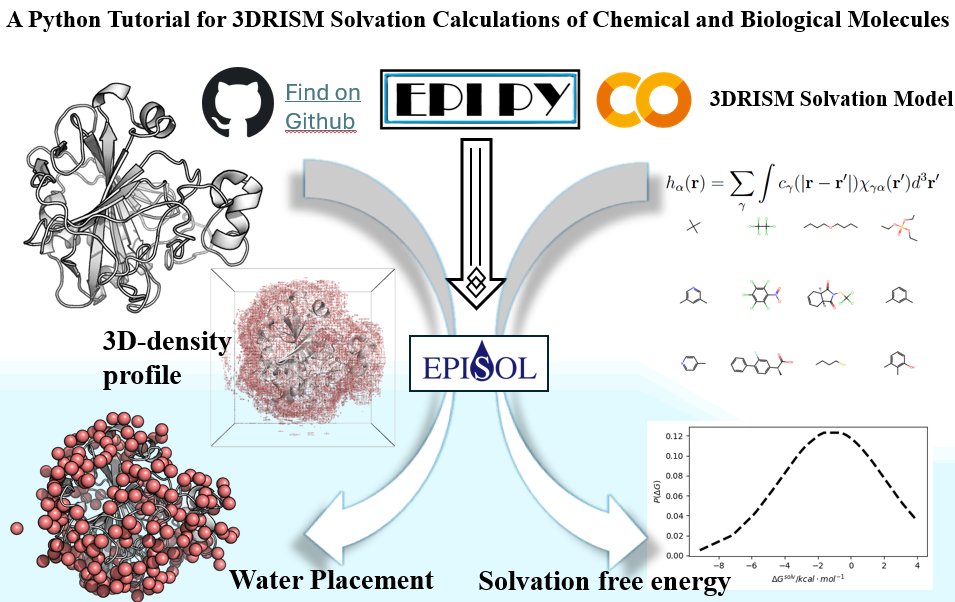
New preprint! Using python interface to perform solvation calculations for molecules both small and large!
chemrxiv.org/engage/chemrxiv…
7
28
2,026
Yiannis Galdadas retweeted
6 Aug 2025
Accelerating Protein Design by Scaling Experimental Characterization
🚀 New preprint from David Baker!🚀
1. This preprint introduces a novel workflow called Semi-Automated Protein Production (SAPP) that significantly accelerates the experimental validation of de novo protein designs. SAPP enables rapid, modular, and cost-effective protein production and characterization, allowing for the purification and analysis of hundreds of protein designs per day.
2. The SAPP protocol leverages a standardized cloning approach and optimized purification pipeline to achieve at least a tenfold increase in throughput compared to traditional methods. It reduces the total hands-on time to just 6 hours for end-to-end execution, making it highly efficient for large-scale protein design campaigns.
3. A key innovation is the use of a background-suppressing cassette in the cloning process, which eliminates the need for colony isolation and sequencing, thus bypassing traditional multi-day cloning protocols. This approach ensures that the correct clone is the dominant construct in most cases, with a clonal purity of over 90% in many instances.
4. The authors also developed a scalable demultiplexing protocol (DMX) to further reduce costs. DMX converts DNA oligo pools into sequence-verified clonal constructs, enabling the purification and characterization of over 1000 designs at a cost of $5 per construct. This protocol integrates seamlessly with SAPP and is particularly useful for large design campaigns.
5. The SAPP and DMX protocols have been successfully applied to characterize tens of thousands of de novo designed proteins, including mini-protein binders, enzymes, and large protein assemblies. These workflows are designed to be widely adoptable, making large-scale experimental testing more accessible and affordable.
6. The integration of these protocols with computational protein design methods is expected to drive the development of new protein design models informed by experimental data. This could also enable active learning approaches, where experimental feedback is used to iteratively improve protein designs.
💻Code: github.com/bwicky/SAPP_DMX
📜Paper: biorxiv.org/content/10.1101/…
#ProteinDesign #ExperimentalValidation #SAPP #DMX #ComputationalBiology #HighThroughput #CostEffective #ActiveLearning

1
45
140
12,019
Yiannis Galdadas retweeted
27 Jul 2025
In Science, researchers present BioEmu—a new #AI model that rapidly and accurately predicts the full range of shapes a protein can adopt, offering a faster, cheaper alternative to traditional molecular simulations.
Learn more: scim.ag/4kVfrWS
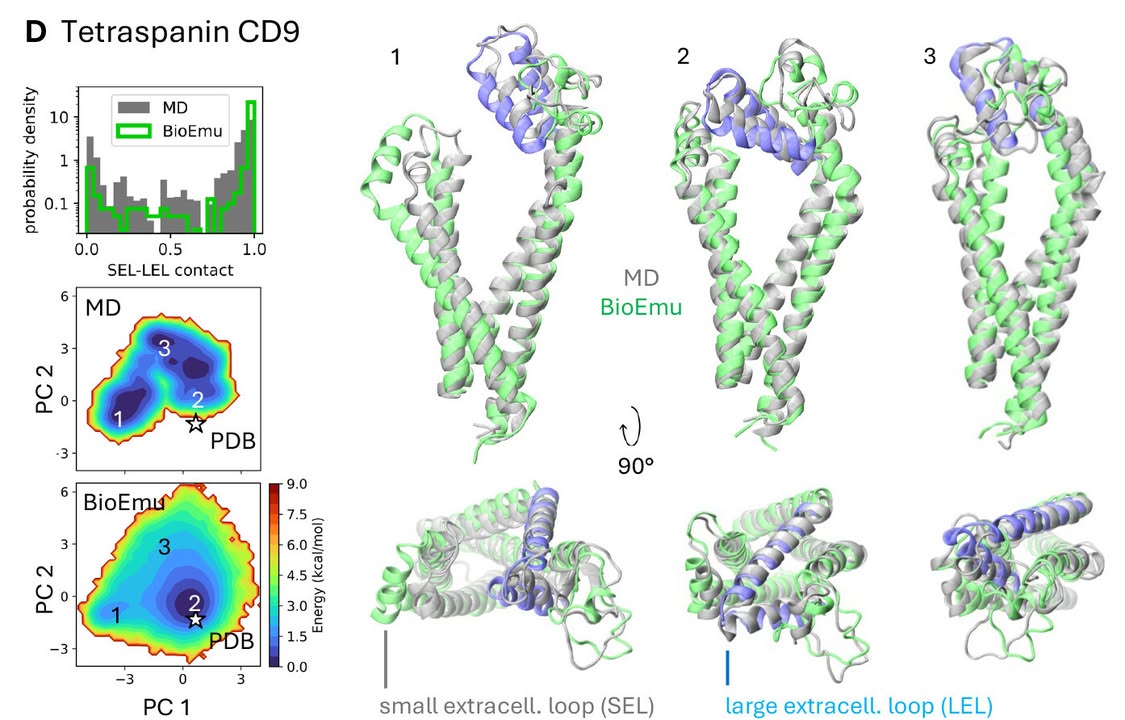
15
87
287
41,381
Yiannis Galdadas retweeted

24 Jul 2025
Thrilled to share our new paper in @ScienceMagazine! 💥
We developed a computational platform to design tiny 'minibinders' from scratch that can guide T cells to kill cancer cells. This is proof-of-concept for targeting intracellular cancer antigens with a new modality! 🎯
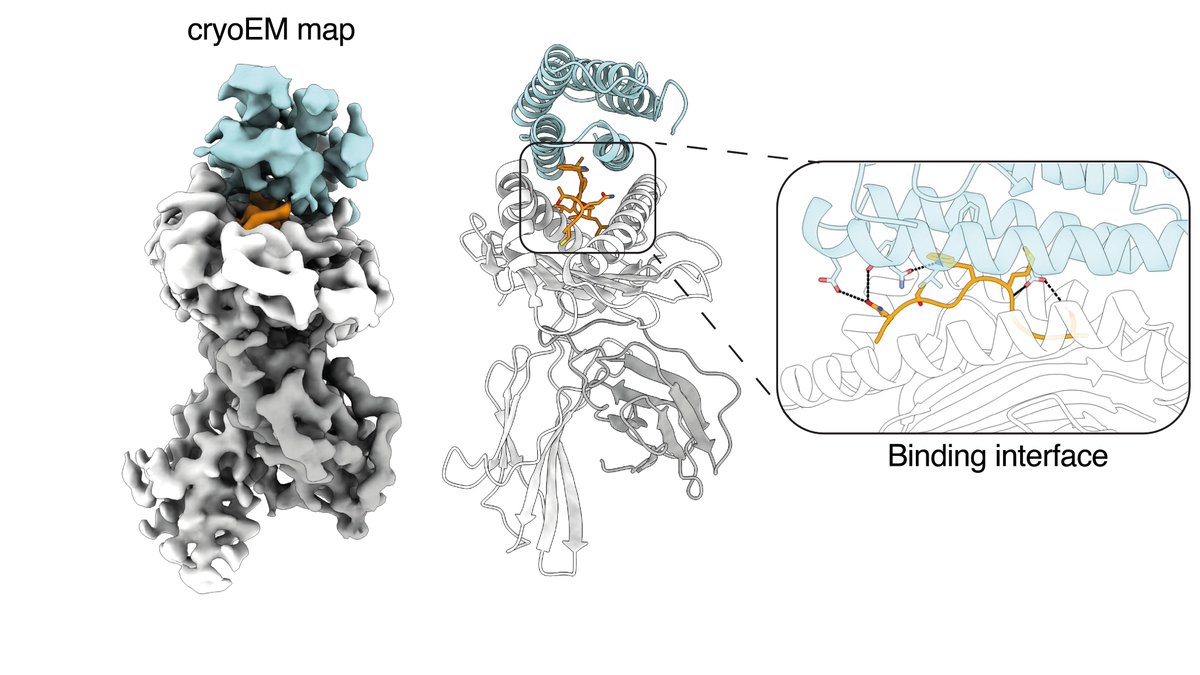
ALT CryoEM map of NY-ESO-1/HLA-A*02:01-bound minibinder
5
48
279
21,763