

May 31

There is a great deal of valuable nuance here regarding the importance of prior knowledge. This section especially stood out to me given my recent experiences with pre-questions, their potential to prime prior knowledge, and their ability to direct and sustain attention.
open.substack.com/pub/solint…

1
62

Mar 14
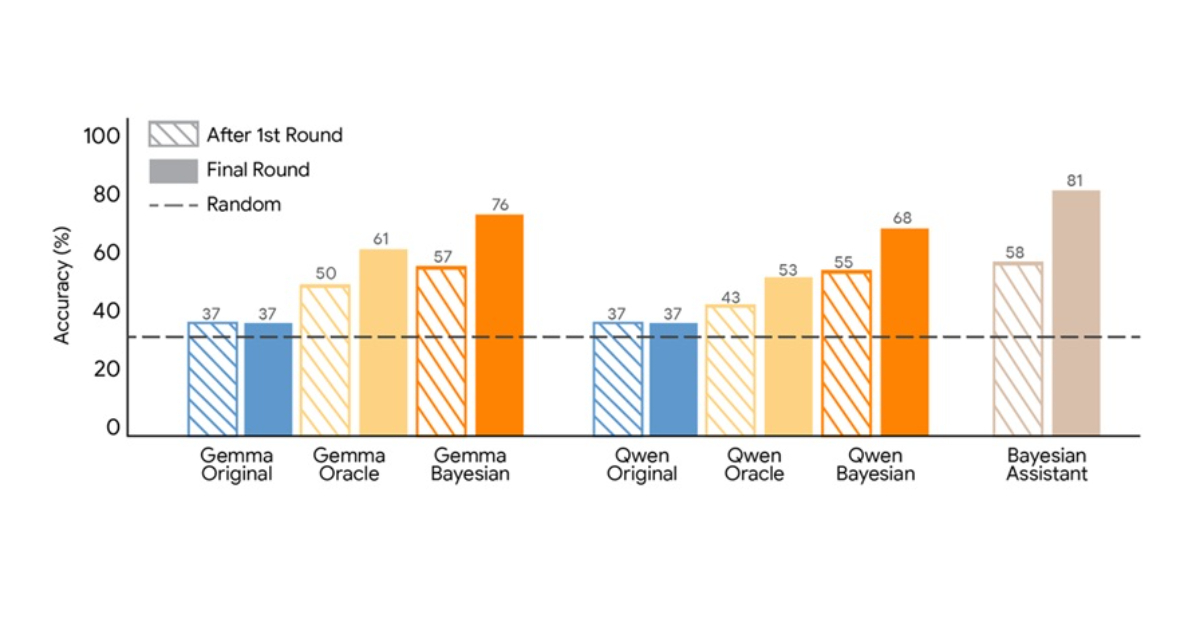
Google just flipped how LLMs learn with Bayesian Teaching—basically, teaching models by optimizing the data they get, not just feeding tons of random info. They’re using probabilistic reasoning to pick the *best* training examples, which could seriously cut down training costs and boost model precision. This isn’t just smarter training; it’s a new mindset for AI education. 🤯
If this scales, we might see LLMs that learn faster and generalize better without gargantuan datasets. Imagine AI that *knows what to learn next* instead of just absorbing everything blindly. Game changer for efficiency and alignment. Thoughts on Bayesian methods taking over model training?
news.google.com/rss/articles…
#GoogleAI #BayesianLearning #LargeLanguageModels
6

Jan 27
💹 Can AI Accurately Forecast Metal Prices?
A new study uses Gaussian Process Regression (GPR) optimized with Bayesian learning to forecast silver prices — achieving an astounding 99.97% correlation accuracy across 13 years of daily market data. 🤖📊
🧩 The Problem
Commodity price volatility makes traditional econometric models like ARIMA and VAR struggle with noisy, nonlinear data. Investors and policymakers often face uncertainty when planning around price fluctuations. ⚖️📉
This study bridges that gap with an AI-powered, probabilistic model built to learn uncertainty itself. 🎯
⚙️ Here’s How It Works
🔹 Gaussian Process Regression (GPR) → builds a probabilistic model that captures nonlinear trends and uncertainty in time-series data
🔹 Bayesian Optimization → fine-tunes parameters for maximum accuracy and minimal overfitting
🔹 Cross-Validation Framework → tests model robustness across 13 years (2011–2024) of global silver prices
📊 Key Results
✅ Relative RMSE: 0.2257%
✅ MAE: 0.0389
✅ Correlation Coefficient: 99.967%
✅ Forecast Window: 13 years
⚡ The model surpasses traditional forecasts in precision and adaptability, setting a new benchmark for AI-driven econometrics.
🌍 Why It Matters
• 📈 Supports economic planning through accurate commodity forecasts
• 💰 Helps investors navigate volatility with real-time predictive insights
• 🧮 Advances AI-based econometrics using uncertainty modeling
• 🔬 Shows how Bayesian methods improve learning stability in financial systems
🎯 Who It’s For
• Quantitative finance researchers → developing next-gen forecasting models
• Economists & data scientists → analyzing long-term commodity behavior
• Policymakers → reducing uncertainty in fiscal decisions
• AI researchers → exploring Bayesian learning for real-world applications
• Traders → improving price prediction for strategic investments
📘 Read the full study and recommend the Journal of Uncertain Systems to your library today:
👉 doi.org/10.1142/S17528909245…
This study proves that machine learning Bayesian optimization can transform financial forecasting — making it more reliable, interpretable, and data-driven.
AI isn’t just predicting prices — it’s redefining how we understand uncertainty itself. 🌐✨
@WorldScienceU @IEEEorg @FintechInsiders @MIT_CSAIL @OpenAI @GoogleDeepMind @arxiv @QuantInsti @ForbesScience @BloombergAsia @ReutersBiz @TheEconomist @CNBC @Investopedia @DataScienceCtrl
#GaussianProcess #BayesianOptimization #AIinFinance #QuantFinance #TimeSeriesForecasting #Econometrics #MachineLearning #SilverPriceForecast #FinancialModeling #BayesianLearning #PredictiveAnalytics #AIResearch #EconomicAI #QuantAnalysis #WorldScientific #DataScience

1
163

19 Nov 2025
Bayesian Learning for Accurate and Robust Biomolecular Force Fields
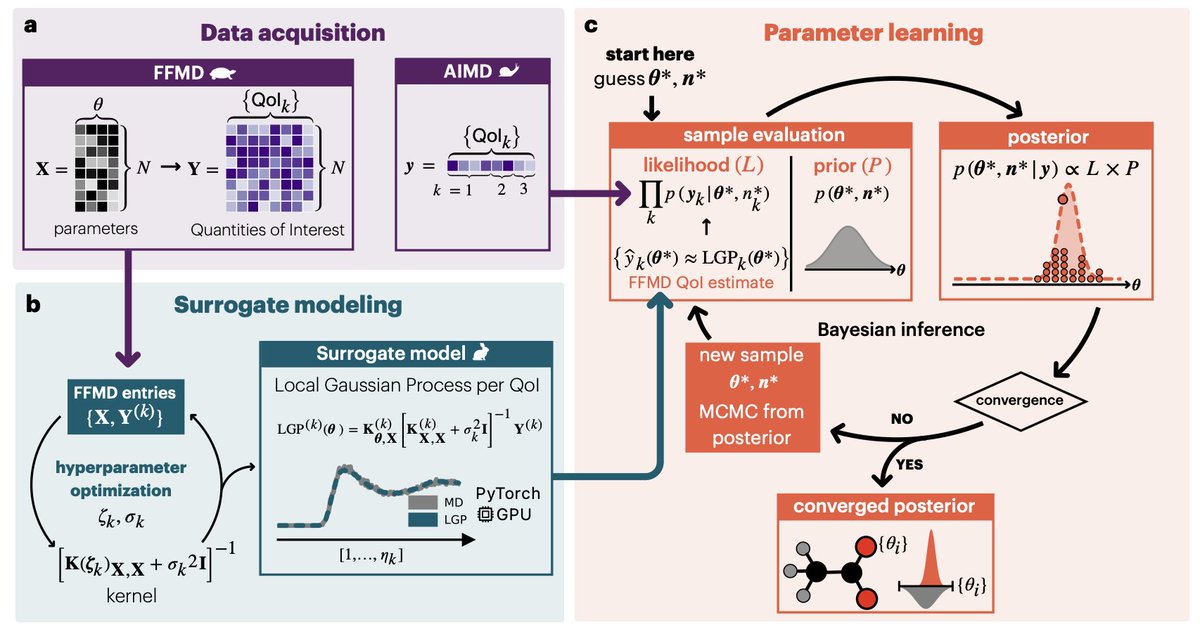
1. A new Bayesian framework has been introduced to optimize biomolecular force fields using ab initio molecular dynamics (AIMD) data. This method provides a transparent, data-driven approach to develop accurate and transferable force fields for complex biological systems.
2. The core innovation lies in the probabilistic representation of model parameters and data, allowing for the natural emergence of uncertainty and transferability. This approach yields interpretable models and enhances confidence in computational descriptions of biophysical systems.
3. The study demonstrates the method using 18 biologically relevant molecular fragments, including proteins, nucleic acids, and lipids. The optimized parameters show significant improvements over existing force fields, particularly for charged systems.
4. A key application is the simulation of calcium binding to cardiac troponin, a critical event in cardiac contraction. The Bayesian approach provides multiple parameter sets, offering insights into the robustness and variability of the model predictions.
5. The method leverages Local Gaussian Processes (LGP) to accelerate Bayesian inference, making it computationally feasible for complex systems. This integration of machine learning and Bayesian statistics offers a powerful tool for force field development.
6. The study highlights the transferability of optimized parameters from small fragments to larger biomolecular systems, validated through simulations of aqueous solutions and protein-ligand interactions. This demonstrates the broad applicability of the framework.
📜Paper: arxiv.org/abs/2511.05398v1
#BayesianLearning #MolecularDynamics #ForceFields #Biophysics #ComputationalBiology
3
18
1,175

31 Oct 2025
Probability Theory Linear Algebra Uncertainty Modeling = Statistical/BayesianLearning
5
30 Sep 2025
RAPTOR-GEN: RApid PosTeriOR GENerator for Bayesian Learning in Biomanufacturing
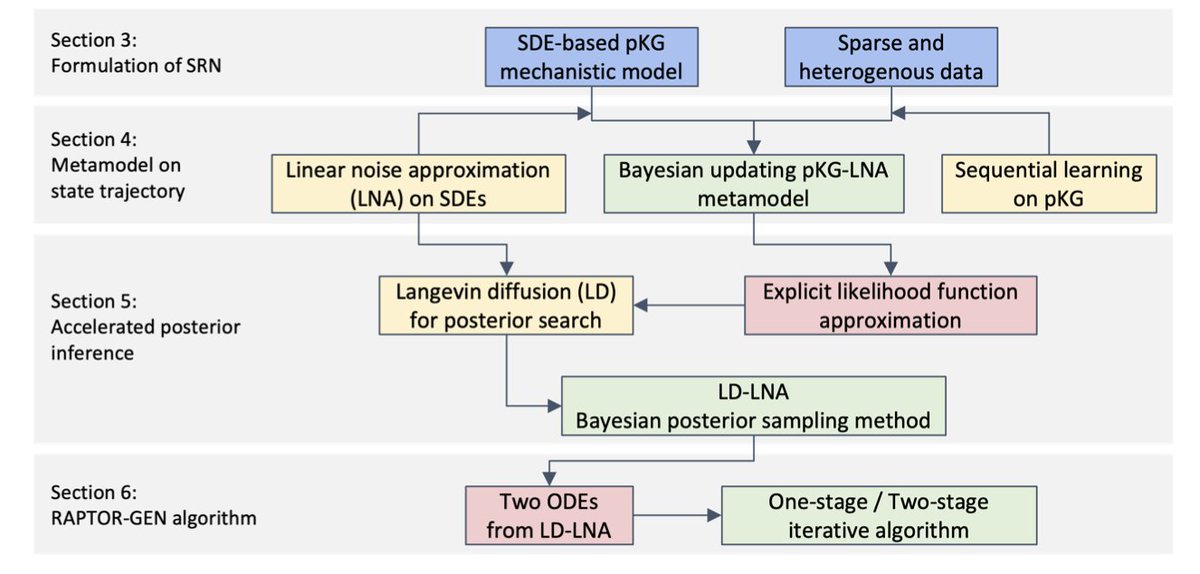
1. RAPTOR-GEN is a novel Bayesian learning framework designed to accelerate intelligent digital twin development in biomanufacturing, addressing the challenges of sparse and heterogeneous data. It leverages a multi-scale probabilistic knowledge graph (pKG) to capture the nonlinear dynamics of bioprocesses.
2. The framework consists of two key components: an interpretable metamodel integrating linear noise approximation (LNA) that exploits the structural information of bioprocessing mechanisms, and an efficient Bayesian posterior sampling method using Langevin diffusion (LD) to accelerate posterior exploration.
3. RAPTOR-GEN generalizes the LNA approach to circumvent the challenge of step size selection, facilitating robust learning of mechanistic parameters with provable finite-sample performance guarantees. It provides a fast and robust algorithm with controllable error.
4. Numerical experiments demonstrate the effectiveness of RAPTOR-GEN in uncovering the underlying regulatory mechanisms of biomanufacturing. It shows superior performance in parameter inference and computational efficiency compared to traditional methods like ULA and ABC-SMC.
5. The study highlights the potential of RAPTOR-GEN to advance scientific understanding of bioprocess regulatory mechanisms and support the development of flexible, automatic, and robust biomanufacturing systems.
📜Paper: arxiv.org/abs/2509.20753
#Biomanufacturing #BayesianLearning #DigitalTwin #ProbabilisticModeling #ComputationalBiology
642
20 Aug 2025
BLIPs: Bayesian Learned Interatomic Potentials
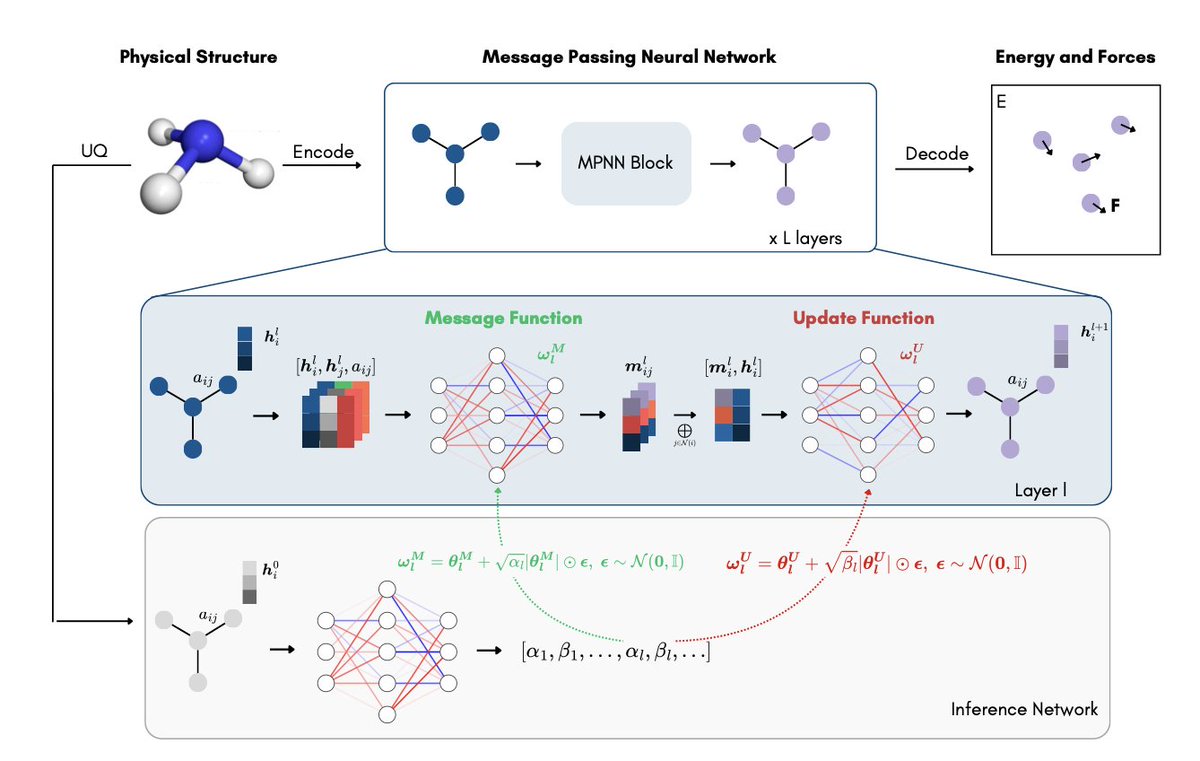
1. A new method called BLIPs (Bayesian Learned Interatomic Potentials) has been proposed to address the limitations of Machine Learning Interatomic Potentials (MLIPs) in handling out-of-distribution data and providing uncertainty estimates. This is a significant advancement in simulation-based chemistry, where accurate predictions and reliable uncertainty quantification are crucial.
2. BLIPs introduce a scalable, architecture-agnostic variational Bayesian framework that can be applied to train or fine-tune MLIPs. The key innovation is the use of an adaptive version of Variational Dropout, which allows the model to capture input-dependent uncertainty by injecting Gaussian noise into the weights of the message and update functions of Message Passing Neural Networks (MPNNs).
3. The method is particularly effective in data-scarce and out-of-distribution regimes, which are common challenges in computational chemistry. Empirical results show that BLIPs not only improve predictive accuracy but also provide well-calibrated uncertainty estimates, outperforming standard MLIPs and other uncertainty quantification methods like Deep Ensembles and MC Dropout.
4. BLIPs integrate seamlessly with equivariant message-passing architectures, maintaining the essential physical symmetries of atomic-scale systems. This integration ensures that the model remains suitable for modeling complex interactions while providing uncertainty quantification without compromising on accuracy.
5. The computational overhead of BLIPs is minimal compared to deterministic models, making it a practical solution for large-scale simulations. Fine-tuning pretrained MLIPs with BLIPs consistently enhances performance and provides reliable uncertainty estimates, demonstrating the method's potential for improving existing models.
6. The effectiveness of BLIPs is demonstrated through various simulation-based chemistry tasks, including modeling the dynamics of charged particles, learning interatomic potentials for ammonia molecules, and fine-tuning pretrained models on silica glass structures. These experiments highlight BLIPs' ability to handle both small and large systems with high accuracy and reliable uncertainty quantification.
7. The code accompanying this work is available at github.com/dario-coscia/blip, allowing researchers to easily implement and experiment with BLIPs in their own projects. This open-source approach promotes further research and development in the field of machine learning for computational chemistry.
📜Paper: arxiv.org/abs/2508.14022
#BLIPs #BayesianLearning #MachineLearningInteratomicPotentials #ComputationalChemistry #UncertaintyQuantification #SimulationBasedChemistry
1
6
27
1,591


17 Jun 2025
If we want AGI, it may have to learn like a child, and be raised like one.
linkedin.com/feed/update/urn…
#AI #ArtificialIntelligence #ChildDevelopment #CognitiveScience #Exploration #LearningToLearn #Empowerment #CausalReasoning #BayesianLearning #AIAlignment #AlisonGopnik #Oxford
17

8 Jun 2025
Testing an Active Inference agent in a 3D drone navigation task — it infers its own position and velocity from noisy observations, and selects actions by rolling out belief-based trajectories and minimizing expected free energy.
#ActiveInference #BayesianLearning #AI #AGI
1
3
304

1 Jun 2025
Great discussion with Risana Zitha on my upcoming book — co-authored with Tsakane Mongwe and Rendani Mbuvha — exploring Bayesian machine learning in qualitative finance. Bridging rigorous AI methods with nuanced financial insight. #AI #Finance #BayesianLearning #UNU amazon.co.uk/Bayesian-Machin…

7
36
1,311
27 May 2025
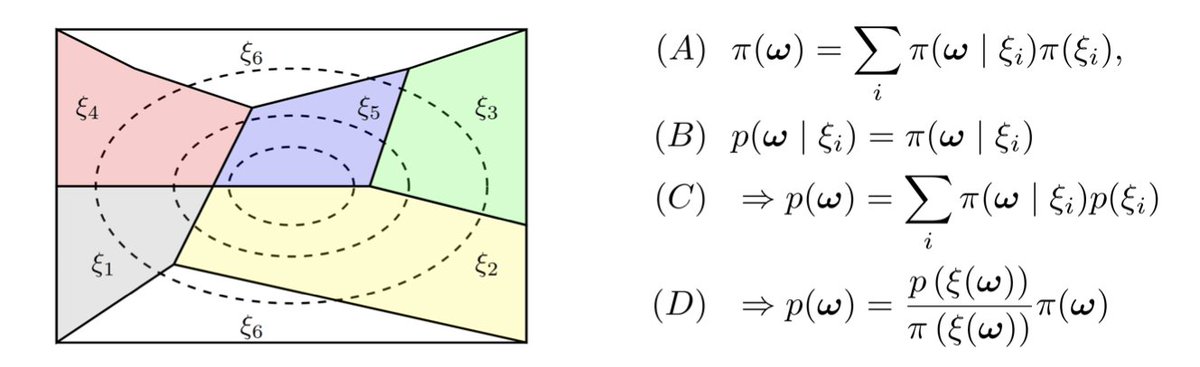
Unfolding AlphaFold’s Bayesian Roots in Probability Kinematics
1.This theoretical paper reinterprets AlphaFold1 (AF1)’s potential energy function as an instance of probability kinematics (PK), a principled Bayesian update mechanism for incorporating uncertain evidence—offering a rigorous alternative to its original justification via physical potentials of mean force (PMFs).
2.Unlike AlphaFold2 and AlphaFold3, AF1 minimizes a learned potential function parameterized over dihedral angles and pairwise distances. The authors argue this potential should not be seen as thermodynamic but as the result of a generalized Bayesian update using PK.
3.PK—also known as Jeffrey conditioning—updates a prior distribution based on revised probabilities over a partition, without conditioning on a specific observed event. It allows AF1’s prior over angles to be updated by "soft" evidence from deep-learning-predicted distance distributions.
4.The paper constructs a probabilistic framework tailored to AF1, showing how its three main potential terms (dihedral, distance, and reference potentials) fit into the PK update formula. The final AF1 potential corresponds to a maximum a posteriori (MAP) estimate under this model.
5.Unlike PMFs derived from Boltzmann distributions over physical systems, AF1's potential reflects updates of empirical priors based on distance evidence. The paper demonstrates that PK justifies knowledge-based potentials (KBPs) and that AF1’s form is a compositional Bayesian update.
6.The authors derive a formal PK update equation suitable for infinite partitions and continuous variables (e.g., distributions over protein dihedral angles and distances), and show how this framework allows principled probabilistic modeling of biomolecular structure.
7.To illustrate PK in a controlled setting, they introduce a synthetic 2D model where a von Mises angular prior (analogous to AF1's dihedral angle prior) is updated with evidence on Euclidean distances, achieving precise posterior recovery validated via hypothesis testing.
8.The synthetic experiment shows that omitting the PK reference term leads to poor posterior estimates, reinforcing the theoretical necessity of the reference distribution—a key aspect missing from naïve knowledge-based or PMF analogies.
9.The authors empirically confirm the validity of the J-condition (p(angles|distances) ≈ π(angles|distances)) using AF1-generated samples, justifying the use of PK to update local priors without retraining or redefining likelihoods.
10.This reinterpretation highlights AF1 as an early example of a compositional probabilistic deep learning model, where empirical Bayes priors are refined with learned evidence—a strategy with broad potential for future structural biology models.
11.The paper suggests that PK can serve as a foundation for combining simple probabilistic components (e.g., priors on angles, distances, and energies) into deep models, offering a path toward interpretable and generalizable generative frameworks in computational biology.
12.More broadly, the authors call for revisiting foundational assumptions in structural bioinformatics and view AF1’s success as evidence of the power of principled Bayesian updates even in high-dimensional deep learning systems.
📜Paper: arxiv.org/abs/2505.19763
#AlphaFold #BayesianLearning #ProteinFolding #ProbabilityKinematics #AF1 #GenerativeBiology #DeepLearning #CompositionalModels #StructuralBioinformatics #KnowledgeBasedPotentials
3
19
1,287
27 May 2025
Unfolding AlphaFold’s Bayesian Roots in Probability Kinematics
1.This theoretical paper reinterprets AlphaFold1 (AF1)’s potential energy function as an instance of probability kinematics (PK), a principled Bayesian update mechanism for incorporating uncertain evidence—offering a rigorous alternative to its original justification via physical potentials of mean force (PMFs).
2.Unlike AlphaFold2 and AlphaFold3, AF1 minimizes a learned potential function parameterized over dihedral angles and pairwise distances. The authors argue this potential should not be seen as thermodynamic but as the result of a generalized Bayesian update using PK.
3.PK—also known as Jeffrey conditioning—updates a prior distribution based on revised probabilities over a partition, without conditioning on a specific observed event. It allows AF1’s prior over angles to be updated by "soft" evidence from deep-learning-predicted distance distributions.
4.The paper constructs a probabilistic framework tailored to AF1, showing how its three main potential terms (dihedral, distance, and reference potentials) fit into the PK update formula. The final AF1 potential corresponds to a maximum a posteriori (MAP) estimate under this model.
5.Unlike PMFs derived from Boltzmann distributions over physical systems, AF1's potential reflects updates of empirical priors based on distance evidence. The paper demonstrates that PK justifies knowledge-based potentials (KBPs) and that AF1’s form is a compositional Bayesian update.
6.The authors derive a formal PK update equation suitable for infinite partitions and continuous variables (e.g., distributions over protein dihedral angles and distances), and show how this framework allows principled probabilistic modeling of biomolecular structure.
7.To illustrate PK in a controlled setting, they introduce a synthetic 2D model where a von Mises angular prior (analogous to AF1's dihedral angle prior) is updated with evidence on Euclidean distances, achieving precise posterior recovery validated via hypothesis testing.
8.The synthetic experiment shows that omitting the PK reference term leads to poor posterior estimates, reinforcing the theoretical necessity of the reference distribution—a key aspect missing from naïve knowledge-based or PMF analogies.
9.The authors empirically confirm the validity of the J-condition (p(angles|distances) ≈ π(angles|distances)) using AF1-generated samples, justifying the use of PK to update local priors without retraining or redefining likelihoods.
10.This reinterpretation highlights AF1 as an early example of a compositional probabilistic deep learning model, where empirical Bayes priors are refined with learned evidence—a strategy with broad potential for future structural biology models.
11.The paper suggests that PK can serve as a foundation for combining simple probabilistic components (e.g., priors on angles, distances, and energies) into deep models, offering a path toward interpretable and generalizable generative frameworks in computational biology.
12.More broadly, the authors call for revisiting foundational assumptions in structural bioinformatics and view AF1’s success as evidence of the power of principled Bayesian updates even in high-dimensional deep learning systems.
📜Paper:
arxiv.org/abs/2505.19763
#AlphaFold #BayesianLearning #ProteinFolding #ProbabilityKinematics #AF1 #GenerativeBiology #DeepLearning #CompositionalModels #StructuralBioinformatics #KnowledgeBasedPotentials
1
3
6
1,761

21 May 2025
🤖 What if your AI could explain its uncertainty?
Unpack how modern agents use MCP, entropy signals, and probabilistic modeling to make informed, auditable decisions 🧩📊
👉 medium.com/@rogt.x1997/entro…
#ResponsibleAI #CognitiveArchitecture #BayesianLearning #LangGraph #MCPAgents
19
27 Apr 2025
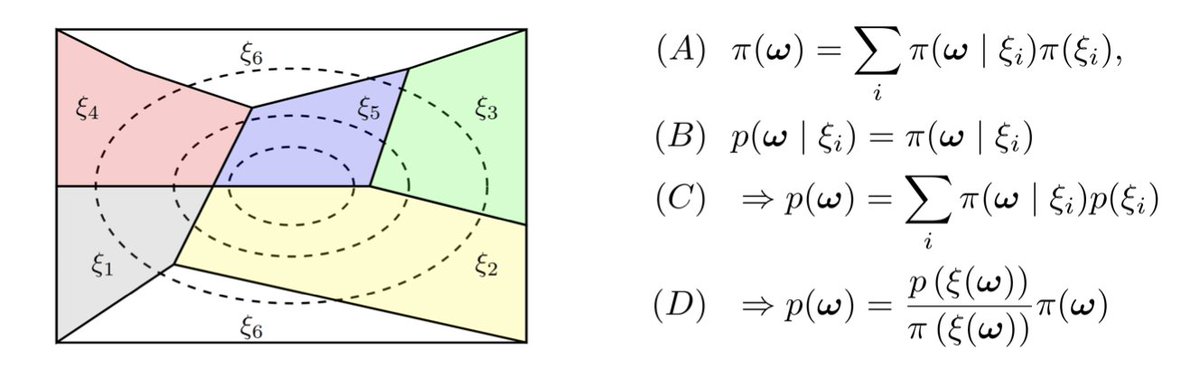
Molecular property prediction using pretrained-BERT and Bayesian active learning: a data-efficient approach to drug design @BMCBiology
1. This study integrates pretrained BERT (MolBERT) with Bayesian active learning (AL) to significantly improve drug discovery by efficiently predicting molecular properties with minimal labeled data. This method enhances data efficiency and accelerates drug design processes.
2. The integration of BERT, pretrained on 1.26 million compounds, allows the model to leverage rich molecular representations that enable better uncertainty estimation, a key factor for active learning in drug discovery. This separates representation learning from uncertainty estimation, optimizing both.
3. Using Bayesian active learning, the study identifies the most informative compounds for labeling, improving model performance while reducing the number of iterations needed. In comparison to conventional AL, the proposed method achieves the same results with 50% fewer iterations.
4. The experiments, including toxic compound prediction on the Tox21 and ClinTox datasets, demonstrate that the BERT-based approach outperforms traditional methods, with better model calibration and faster convergence in sample selection, particularly in toxicology.
5. The study also compares the performance of various acquisition functions, including Expected Predictive Information Gain (EPIG) and Bayesian Active Learning by Disagreement (BALD), showing that EPIG consistently provides more stable and reliable results with the BERT representations.
6. Visualizations such as UMAP and PCA illustrate the superior ability of BERT representations to create a more structured embedding space, facilitating faster and more accurate identification of useful molecular features in the active learning process.
7. One key takeaway is that using high-quality molecular representations, like those generated by MolBERT, drastically enhances uncertainty estimation, allowing for more reliable selection of compounds even when starting with limited data.
8. This work paves the way for more efficient and scalable drug discovery workflows, particularly in early-stage compound prioritization and toxicity prediction, making it a valuable tool for pharmaceutical applications.
💻Code: github.com/Arslan-Masood/Act…
📜Paper: jcheminf.biomedcentral.com/a…
#DrugDiscovery #AIinBiology #MachineLearning #ActiveLearning #Bioinformatics #DrugDesign #MolecularPrediction #BayesianLearning #BERT #Chemoinformatics

4
36
2,718
27 Apr 2025
Molecular property prediction using pretrained-BERT and Bayesian active learning: a data-efficient approach to drug design @BMCBiology
1. This study integrates pretrained BERT (MolBERT) with Bayesian active learning (AL) to significantly improve drug discovery by efficiently predicting molecular properties with minimal labeled data. This method enhances data efficiency and accelerates drug design processes.
2. The integration of BERT, pretrained on 1.26 million compounds, allows the model to leverage rich molecular representations that enable better uncertainty estimation, a key factor for active learning in drug discovery. This separates representation learning from uncertainty estimation, optimizing both.
3. Using Bayesian active learning, the study identifies the most informative compounds for labeling, improving model performance while reducing the number of iterations needed. In comparison to conventional AL, the proposed method achieves the same results with 50% fewer iterations.
4. The experiments, including toxic compound prediction on the Tox21 and ClinTox datasets, demonstrate that the BERT-based approach outperforms traditional methods, with better model calibration and faster convergence in sample selection, particularly in toxicology.
5. The study also compares the performance of various acquisition functions, including Expected Predictive Information Gain (EPIG) and Bayesian Active Learning by Disagreement (BALD), showing that EPIG consistently provides more stable and reliable results with the BERT representations.
6. Visualizations such as UMAP and PCA illustrate the superior ability of BERT representations to create a more structured embedding space, facilitating faster and more accurate identification of useful molecular features in the active learning process.
7. One key takeaway is that using high-quality molecular representations, like those generated by MolBERT, drastically enhances uncertainty estimation, allowing for more reliable selection of compounds even when starting with limited data.
8. This work paves the way for more efficient and scalable drug discovery workflows, particularly in early-stage compound prioritization and toxicity prediction, making it a valuable tool for pharmaceutical applications.
💻Code: github.com/Arslan-Masood/Act…
📜Paper: jcheminf.biomedcentral.com/a…
#DrugDiscovery #AIinBiology #MachineLearning #ActiveLearning #Bioinformatics #DrugDesign #MolecularPrediction #BayesianLearning #BERT #Chemoinformatics
1
755

New lens on governance: Learning model shows directors matter and drive 10% of volatility when appointed. Study reveals when investors expect directors to have greater impact. #BayesianLearning #Boards #GenerationalDiversity #KnowledgeCapital jfqa.org/wp-content/uploads/…

298

7 Apr 2025
New open access paper in Automatica: Bayesian online learning for human-assisted target localization. Fusing human sketches with sensor data to improve tracking, even with limited info. Check it out: doi.org/10.1016/j.automatica… #HumanRobotInteraction #BayesianLearning #TargetTracking

1
5
809

26 Feb 2025
#Mathematics #callforreading
📝 "Bayesian Learning in an Affine #GARCH Model with Application to #PortfolioOptimization"
✍️ By Technical University of Munich researchers et al.
📊 buff.ly/3XCTZOq
🏷️ #Statistics #BayesianLearning
@MDPIOpenAccess @ComSciMath_Mdpi
50