Discriminator-guided Inverse Folding for Multi-property Protein Design
1 DGIF introduces a plug-and-play way to steer an inverse-folding model toward multiple protein properties at once, without fine-tuning the generative model and without needing multi-property-labeled datasets.
2 Core idea: during autoregressive sequence generation, DGIF backpropagates gradients from an auxiliary discriminator into the decoder’s internal history state (KV-cache / “history states”), then re-samples the next residue from the updated distribution—repeating this at every step.
3 The discriminator is a composition of multiple single-property predictors. Each predictor can be trained independently on a dataset labeled for only that property, and DGIF combines their signals with weights (beta_i) to perform multi-objective optimization.
4 DGIF is implemented on top of ESM-IF1, producing three variants: DG-Thermo (thermostability), DG-Sol (solubility), and DG-Dual (thermostability solubility). The base inverse-folding model parameters remain unchanged.
5 For thermostability guidance, the paper trains a ΔΔG predictor using ESM-IF1 representations on the Megascale dataset (≈700k mutation–stability pairs), with additional evaluation on FireProt and S669. The predictor outperforms several classic baselines (e.g., FoldX/Rosetta/Thermonet) and is competitive with ThermoMPNN.
6 DG-Thermo improves design outcomes vs unguided ESM-IF1 on: (i) average top-K recall for stabilizing mutations on Megascale test proteins, and (ii) “success rate” of full-sequence designs that both improve predicted stability (ΔΔG > 1.0 kcal/mol) and maintain foldability (predicted structure RMSD < 2 Å).
7 Mechanistic signals emerge naturally: DG-Thermo-designed proteins show more salt bridges and hydrophobic interactions, and amino-acid composition shifts consistent with thermophilic trends (e.g., increased L/P/R/W and decreased D/K/M/Q), despite these rules not being explicitly encoded as constraints.
8 MD validation: for xylanase at 450 K (100 ns), DG-Thermo variants maintain structure (lower RMSD, higher secondary-structure retention) compared with wild type and an unguided ESM-IF1 design; additional CATH-sampled scaffolds show similar stability gains in MD.
9 Solubility guidance: a binary solubility predictor (ESM-IF1 representations MLP) is trained on Khurana et al. and tested on Chang et al.; DG-Sol improves top-K recall on SoluProtMutDB and increases design success rates under joint criteria (better predicted solubility RMSD < 2 Å). On membrane proteins, DG-Sol designs increase surface polar residue proportion, consistent with higher solubility.
10 Multi-property optimization: DG-Dual jointly applies thermostability and solubility predictors and shifts designs toward the Pareto front (better stability/solubility trade-offs) on CATH redesign tasks. Wet-lab validation on Rhodococcus ruber alcohol dehydrogenase (RrADH) tests 10 DG-Dual-suggested single mutations: all improve solubility; 8/10 increase melting temperature. Examples include A50E (≈2x ELISA solubility signal 2.79 °C Tm) and S223A ( 6.47 °C Tm with concurrent solubility gain).
💻Code: github.com/aweqardf/ESM-IF1-…
📜Paper: doi.org/10.1002/advs.75988
#ProteinDesign #InverseFolding #MultiObjectiveOptimization #ESM #ComputationalBiology #MachineLearning #Thermostability #Solubility #ProteinEngineering

5
23
1,441
Probe Before You Edit: Probing-Guided Molecular Optimization for LLM Agents in Structure-Based Drug Design
1. The paper argues that today’s LLM agents in structure-based drug design (SBDD) often “edit blindly”: they commit to a molecular change before having pocket-specific evidence about how local edits will affect binding affinity and druggability, so single edits rarely improve both at once.
2. To quantify this, the authors introduce two diagnostics for multi-objective optimization steps: (i) how often a single edit achieves joint improvement in affinity and druggability, and (ii) how often improving one objective causes the other to deteriorate (objective interference). These metrics expose a consistent failure mode across both “LLM directly edits molecules” and “LLM planner external executor” pipelines.
3. They define affinity as A(m) = −Vina(m) (higher is better) and druggability as D(m) = QED(m) SA(m) (higher is better), then track per-step deltas (∆A, ∆D). Baselines (MoLLM, CIDD, CIDD MOO, LIDDIA) show that joint improvement is uncommon, while affinity-only or druggability-only gains dominate.
4. The core method, PROBE, is inspired by medicinal chemistry practice: run controlled “analog” perturbations to measure response before committing to a direction. PROBE operationalizes this as “probe before you edit” rather than “edit then observe.”
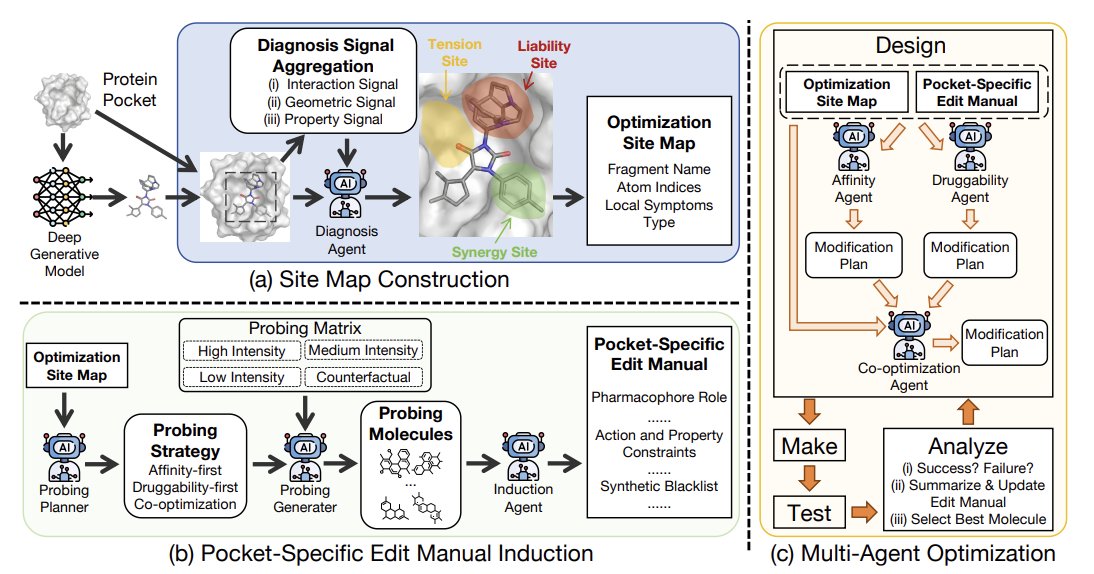
5. PROBE starts by constructing a pocket-specific Site Map. It decomposes the ligand into BRICS fragments and aggregates three signal streams: PLIP interaction signals, geometric fit signals (clashes/voids/mismatch), and property signals (e.g., ligand efficiency and structural alerts). A diagnosis LLM then selects 3–4 editable sites and labels each as SYNERGY (potential win-win), TENSION (likely trade-off), or LIABILITY (must-fix substructure).
6. Next is the probing stage that induces a pocket-specific EditManual. A probing planner proposes three strategies (affinity-first, druggability-first, co-optimization). For each strategy, PROBE generates four probe edits (high/medium/low intensity plus a counterfactual reversal), totaling 12 probes per molecule, then docks/scores them to observe (∆A, ∆D) responses and dose-response patterns (e.g., monotone, saturation, activity cliff).
7. The EditManual distills probe evidence into executable constraints per site: pharmacophore role, allowed/forbidden actions, semantic envelopes (size/polarity/flexibility/shape/charge), and blacklists of features that trigger QED/SA liabilities. It also adds cross-site rules (independent vs mutually exclusive edits), turning empirical probe outcomes into a reusable “local policy” for editing.
8. Optimization is then run as a multi-agent DMTA loop: an affinity agent and a druggability agent propose edits under the same Site Map EditManual, cross-review each other for violations/conflicts, and a co-optimization agent reconciles them into a final edit. Candidate selection uses a joint-improvement-only hypervolume-style score relative to the initial molecule, explicitly discouraging steps that improve only one objective.
9. On CrossDocked2020, PROBE improves both affinity and druggability more consistently than prior LLM-agent pipelines, and it substantially reduces objective interference. In per-step statistics, PROBE’s joint-improvement rate rises to 52.8% (vs < 30% for baselines), and its interference rates drop markedly (around 25–30% vs ~60–88% for baselines depending on intent).
10. Ablations show both components matter: organizing edits via the Site Map improves metrics broadly, but the biggest gains come from a probed (evidence-induced) EditManual rather than an LLM-written manual from prior knowledge alone—supporting the paper’s thesis that pocket-specific measured responses, not just better prompting, are key to multi-objective SBDD editing.
📜Paper: arxiv.org/abs/2606.00555
#ComputationalBiology #DrugDiscovery #SBDD #MolecularOptimization #LLM #AIAgents #Cheminformatics #MedicinalChemistry #MultiObjectiveOptimization
1
1
12
1,437
Don’t Retrain, Just Reuse: Recovering Dual-Target Molecules from Single-Target Diffusion Models
1. The paper asks a practical question in structure-based drug design: instead of retraining or adding sampling-time guidance for dual-target generation, can dual-target ligands be recovered by searching the input noise space of a frozen single-target diffusion model, keeping both parameters and denoising dynamics unchanged?
2. It formalizes dual-target design as constrained multi-objective optimization over searched noise inputs and a final candidate panel, jointly optimizing (i) dual-target affinity with explicit balance, (ii) chemical quality (drug-likeness and synthesizability), and (iii) set-level diversity, under hard feasibility constraints.
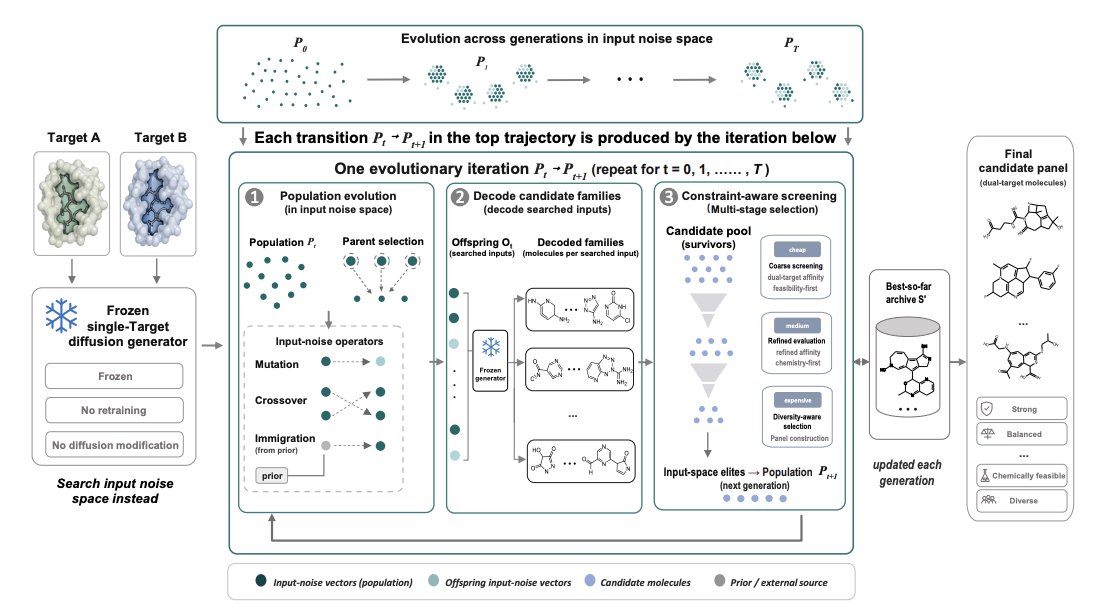
3. The proposed method, REUSE, is a hierarchical evolutionary search in the generator’s input noise space: maintain a population of noise vectors, generate offspring via mutation/crossover/immigration, decode each noise vector into a small “molecular family,” and use family-level evidence (not a single sample) to score and evolve the population toward regions that reliably yield dual-target candidates.
4. A key design choice is family-based fitness: each noise input is evaluated by aggregating scores of the top-ranked molecules within its decoded family, reducing sensitivity to stochastic decoding and favoring noise regions that consistently produce good candidates rather than one-off lucky samples.
5. REUSE uses cost-aware multi-stage environmental selection (coarse-to-fine): cheap docking/chemistry proxies first filter large pools with feasibility-first ranking, then expensive high-fidelity docking is applied only to a reduced frontier; final output is a diverse panel (not a single molecule), with explicit similarity constraints to avoid redundancy.
6. Balance is enforced directly in the affinity objective using a penalty on cross-target score disparity, discouraging “one-target-only” solutions; chemistry control is incorporated via QED/SA-based terms plus hard floors, so optimization does not collapse into unrealistic high-docking-score artifacts.
7. On the Zhou et al. dual-target benchmark (12,917 target pairs, 438 targets), using TargetDiff as the frozen backbone, REUSE achieves the best reported docking-centered dual-target metrics: best average P-2 Vina Dock (-9.26), best Max Vina Dock (-8.64), and highest Dual High Affinity rate (58.3%), improving Dual High Affinity by 20.9 percentage points over the strongest prior baseline (MDRL).
8. Ablations isolate failure modes: removing input-space search sharply degrades dual-target recovery (Dual High Affinity 58.3% → 31.8%); removing balance hurts cross-target consistency; removing chemistry control can slightly improve docking but substantially worsens QED/SA, highlighting why multi-objective constraints matter.
9. The paper provides evidence that the frozen input space has exploitable local structure: high-quality noise “anchors” tend to have enriched high-quality neighbors, supporting why evolutionary local exploration (mutation) can work better than naive random sampling.
📜Paper: arxiv.org/abs/2605.25681
#ComputationalBiology #MachineLearning #DiffusionModels #DrugDiscovery #Polypharmacology #StructureBasedDrugDesign #MolecularGeneration #MultiObjectiveOptimization #EvolutionaryAlgorithms
8
18
1,648
ToolMol: Evolutionary Agentic Framework for Multi-objective Drug Discovery
1. ToolMol addresses a practical failure mode of LLM-based molecule generation: directly emitting SMILES often produces invalid strings (reported >30% invalid even for strong reasoning models), which wastes oracle budget and forces fallbacks to weaker operators in prior work.
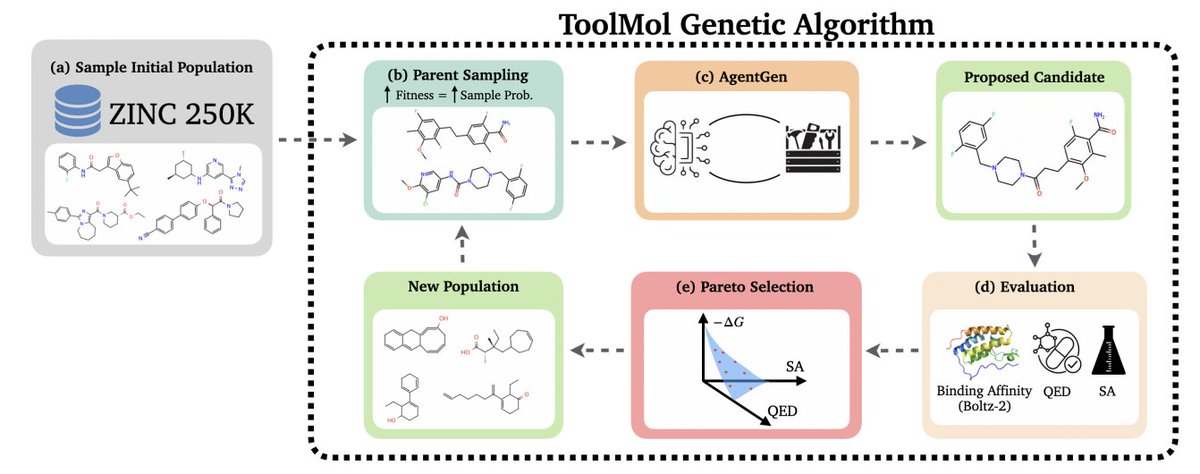
2. The key idea is to stop asking the LLM to “write molecules” and instead let it “edit molecules” via tool-calling: an agentic LLM proposes structured modifications, executed deterministically by RDKit-backed functions, making the final output syntactically valid by construction.
3. Method overview: a multi-objective genetic algorithm maintains a ligand population, selects parents with fitness-proportional sampling, then calls an LLM operator (AgentGen) to perform crossover a few mutations; the next generation is the non-dominated Pareto frontier (binding affinity, QED, SA).
4. The toolbox contains 7 deterministic operations (e.g., crossover molecules, add functional group, replace atom, replace/remove substructure). The LLM supplies parameters (atom indices, groups, bond types), while RDKit enforces valence/graph validity and returns explicit error messages when an operation is impossible.
5. AgentGen is iterative (up to 10 steps; typically fewer). Each tool call updates the conversation with the executed action plus refreshed atom-level structure annotations (substitutable H, ring membership, centrality, etc.) and molecular descriptors (QED, SA, MW, LogP, TPSA, HBD/HBA, rotors).
6. Multi-objective evaluation uses Boltz-2 predicted binding affinity (ΔG), QED (maximize), and SA (minimize). Reporting emphasizes “Filtered Affinity” (top binders that also pass QED > 0.5 and SA < 3.0) plus Pareto hypervolume, reflecting lead-likeness constraints rather than raw affinity alone.
7. Across three targets (c-MET, BRD4, ACAA1), ToolMol ranks best on average and leads on the multi-objective metrics (Filtered Affinity Hypervolume). The paper reports >10% stronger predicted binding affinity than existing methods while producing drug-like and synthesizable candidates.
8. A notable validation step: ToolMol’s top molecules also achieve state-of-the-art Absolute Binding Free Energy (ABFE) results on c-MET and BRD4, improving over MF-LAL by >35% on the reported setup—even though ABFE is not optimized during search (ToolMol optimizes Boltz-2 affinity QED/SA).
9. Ablations isolate why tools matter: swapping ToolMol’s tool-calling operator into MOLLEO’s GA improves results; forcing MOLLEO to retry until valid SMILES reduces invalidity but does not improve (often degrades) optimization metrics, suggesting the gain is not only “validity” but higher-fidelity execution of intended edits.
10. Mechanistic insight from reasoning traces: tool-calling increases concordance between the LLM’s planned chemical changes and the actual applied modifications (ToolMol shows far fewer plan/execution mismatches than direct-SMILES editing), enabling better use of the LLM’s chemical priors during iterative optimization.
📜Paper: arxiv.org/abs/2605.12784
#DrugDiscovery #ComputationalChemistry #Cheminformatics #MolecularDesign #LLM #Agents #ToolCalling #GeneticAlgorithms #MultiObjectiveOptimization #RDKit #ABFE #BindingAffinity
6
37
2,116
Proteinopd: Towards effective and efficient preference alignment for protein design
1 ProteinOPD is a multi-objective preference alignment framework for protein language models (PLMs) that improves target protein properties while preserving “designability” (naturalness/structural plausibility), addressing a common failure mode of post-training alignment: catastrophic forgetting.
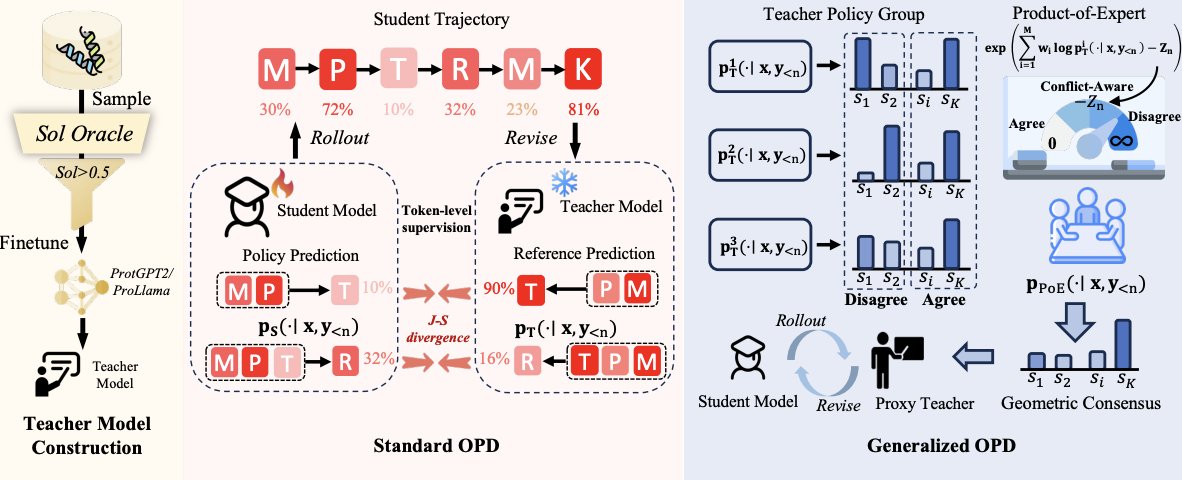
2 The key idea is to adapt a pretrained PLM into several preference-specific teacher models (one per property) using lightweight oracle-guided SFT on a few hundred high-scoring sequences, then distill these teachers into a single shared student using token-level On-Policy Distillation (OPD) on the student’s own rollouts.
3 Why OPD for protein alignment: unlike mode-covering SFT, OPD is mode-seeking and provides dense token-level supervision on the student-visited states, which helps reduce exposure bias and better retains pretrained generative competence compared with directly training on curated “good” sequences.
4 ProteinOPD’s main methodological contribution is extending OPD to multi-objective alignment via a generalized multi-teacher target: at each generation step, it constructs a geometric consensus distribution as a normalized product-of-experts (PoE) over teacher next-token distributions (weighted geometric mean).
5 This PoE consensus is derived as the solution to minimizing the weighted sum of KL divergences from a target distribution q to each teacher, yielding a principled “consensus over coverage” target that emphasizes tokens jointly supported across objectives rather than tokens favored by any single objective.
6 A stability insight: the PoE normalization term Zn is always ≤ 0, and −Zn serves as a free, token-level disagreement/conflict measure among teachers; when objectives conflict strongly, the consensus signal is bounded, improving training stability under antagonistic or noisy preferences.
7 Experimental setup spans unconditional generation (ProtGPT2) and conditional family-guided generation (ProLLaMA on a lysozyme-like superfamily). Designability is evaluated with PPL, novelty vs UniProtKB/ training sets, and ProTrek (conditional semantic consistency). Preference alignment uses oracles: ESMFold-derived pLDDT/PAE (foldability), Protein-Sol (solubility), and TemBERTure (thermostability). Overall trade-offs are summarized by hypervolume (HV).
8 Multi-objective unconditional results: ProteinOPD achieves the best overall trade-off (HV 0.62), outperforming MoMPNN (0.46) and ProGen2 (0.44). Relative to ProtGPT2, it improves pLDDT ( 14.8%), solubility ( 16.9%), and thermostability ( 54.2%), while also improving designability proxies (PPL reduced by 83.7%) and increasing novelty vs training data ( 9.4%).
9 Single-objective analysis shows OPD can transfer property gains from SFT teachers while mitigating SFT-induced novelty/designability loss; in conditional generation it also improves condition fidelity (ProTrek 10% vs Teacher-SFT), consistent with OPD correcting the student on its own visited trajectories rather than forcing coverage of a narrow curated dataset.
10 Efficiency: compared with RL-based alignment (e.g., ProtRL/GRPO-style), ProteinOPD reaches comparable alignment quality with ~8× less training time, benefiting from dense token-level distillation rather than sparse sequence-level rewards and repeated oracle rollouts; teacher construction remains data-efficient (good performance with ~100 filtered sequences, improved further with stricter oracle thresholds).
💻Code: github.com/THU-AI4S/ProteinO…
📜Paper: arxiv.org/abs/2605.10189
#ProteinDesign #ProteinLanguageModels #Alignment #PreferenceLearning #Distillation #MultiObjectiveOptimization #ComputationalBiology #SyntheticBiology #MachineLearning
1
4
29
2,249
MP2D: Constrained Monte Carlo Tree-Guided Diffusion for Multi-Objective Protein Sequence Design
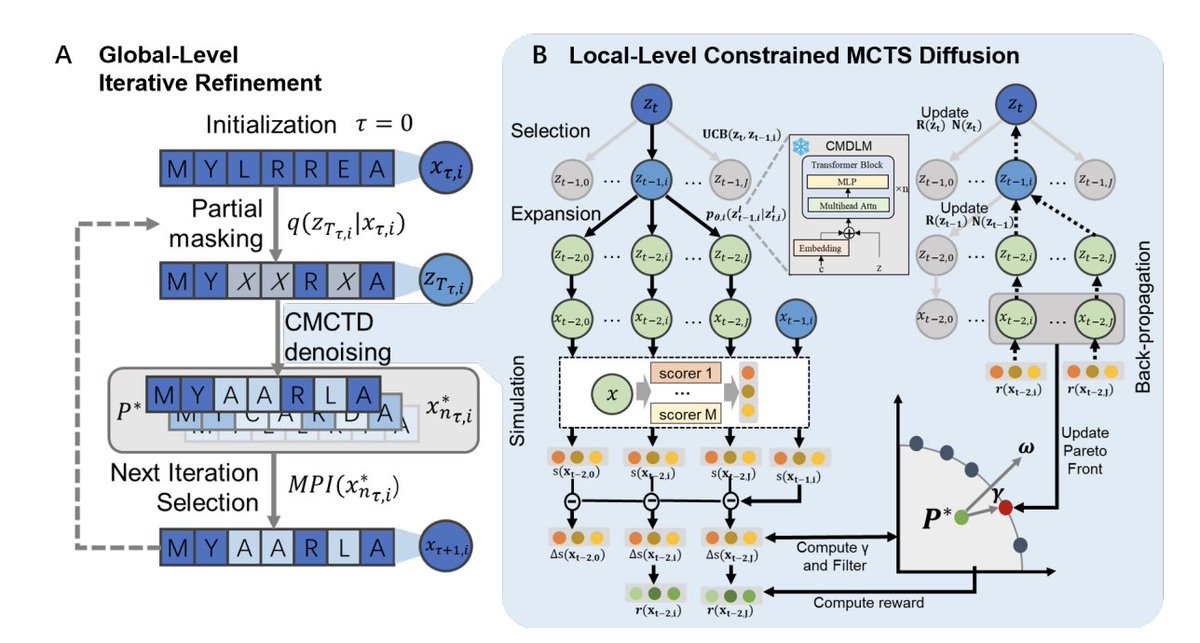
1 MP2D frames diffusion denoising as a constrained sequential decision-making problem and uses Monte Carlo Tree Search (MCTS) to explore many alternative denoising trajectories, rather than committing to a single diffusion path when objectives conflict (e.g., efficacy vs toxicity).
2 The key multi-objective ingredient is Pareto-guided search: each candidate is evaluated by a vector of property predictors, and MCTS selection/expansion is restricted to Pareto non-dominated children, so the search explicitly targets balanced trade-offs instead of relying on fragile scalar weights.
3 To keep Pareto optimization scalable when optimizing 4–5 objectives, MP2D introduces a dynamic Pareto constraint: candidate updates are filtered by alignment to predefined optimization directions (Das–Dennis simplex lattice) using an angular threshold that is adaptively tuned to maintain a stable rejection/acceptance rate, preventing Pareto-front “bloat” and property collapse.
4 MP2D is training-free at optimization time: it plugs in pretrained property evaluators and a pretrained conditional discrete diffusion model, and can swap objectives/predictors without retraining the generative backbone—positioned as a practical workflow for rapidly changing design specifications.
5 The generative backbone is CMDLM, a classifier-free, label-guided conditional masked diffusion language model for peptides (built on an ESM-style transformer). It is pretrained on 2.6M UniProt peptides (length 2–50) and then LoRA fine-tuned for antimicrobial peptides (AMPs) and protein binders (PBs), aiming to narrow the search space to task-relevant sequence regions.
6 CMDLM is evaluated on plausibility and realism using ESM-2 pseudoperplexity, structural foldability via OmegaFold pLDDT, and distributional similarity via Frechet ProtT5 Distance (FPD). Across peptide, AMP, and PB settings, CMDLM is generally more plausible/foldable and closer to target distributions than ProteinGAN, ProtGPT2, and EvoDiff under the paper’s benchmark.
7 The optimization engine (CMCTD) modifies UCB in MCTS by adding a diffusion-posterior–guided exploration term, encouraging exploration that stays close to valid diffusion transitions while still pursuing multi-property reward improvements.
8 MP2D adds a global iterative refinement loop: starting from W seed sequences, it repeatedly partially remasks sequences at random noise levels and reruns constrained MCTS denoising. This is designed to (a) correct early irreversible token decisions typical of single-pass denoising, and (b) increase diversity of optimization routes under noisy global property predictors.
9 On protein binder optimization (5 objectives: hemolysis, non-fouling, solubility, half-life, affinity) for targets 1B8Q and PPP5, MP2D outperforms classical multi-objective optimizers (NSGA-III, SMS-EMOA, SPEA2, MOPSO) and a recent generative baseline (MOG-DFM), achieving lower hemolysis, higher non-fouling/solubility/half-life, and competitive affinity.
10 On AMP optimization (4 objectives: antimicrobial probability, MIC, hemolysis, toxicity), MP2D outperforms AMP-focused multi-objective generative baselines (Multi-CGAN, MPOGAN, HMAMP, MoFormer), improving potency (higher Pamp, lower MIC) while simultaneously reducing safety risks (lower hemolysis and toxicity), addressing the common failure mode where optimizing one property degrades another.
📜Paper: arxiv.org/abs/2605.05829
#ProteinDesign #ComputationalBiology #DiffusionModels #MonteCarloTreeSearch #MultiObjectiveOptimization #AntimicrobialPeptides #PeptideDesign #ProteinEngineering #GenerativeAI #ParetoOptimization
2
27
1,865
RosettaSearch: Multi-Objective Inference-Time Search for Protein Sequence Design
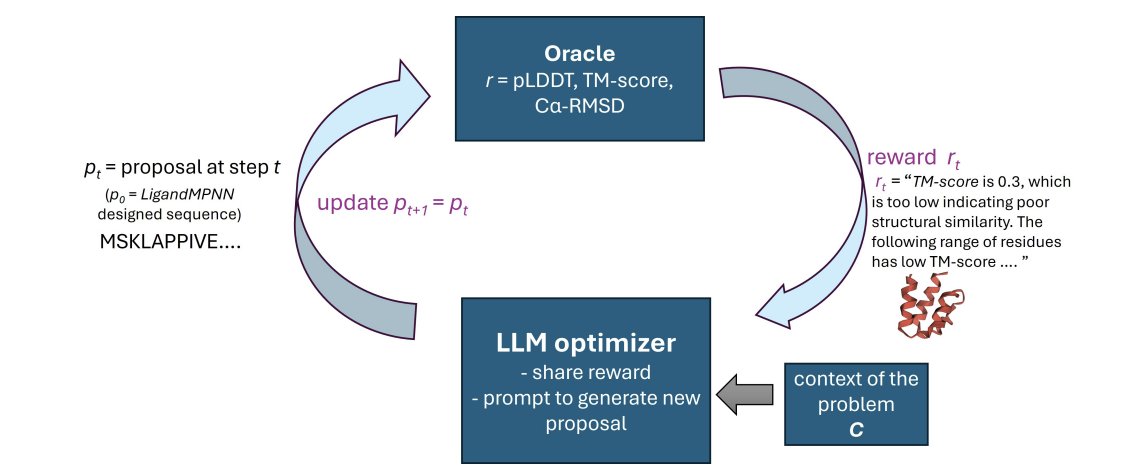
1. RosettaSearch is an inference-time framework that uses LLMs as generative optimizers to refine backbone-conditioned protein sequences via multi-objective search, guided by structural rewards from RosettaFold3, without any retraining or fine-tuning.
2. The key result: on ~400 challenging PDB monomer redesign cases initialized from suboptimal LigandMPNN outputs, RosettaSearch substantially improves structural fidelity (ss-pLDDT, TM-score, Cα-RMSD), yielding about a 2.5× increase in design success rate compared to LigandMPNN single-pass decoding.
3. The method is built around “feedback-driven editing” rather than one-shot generation: the LLM receives (a) global scalar rewards derived from structure prediction metrics and (b) local residue-range annotations highlighting problematic regions (low confidence or high deviation), then proposes targeted sequence edits.
4. Search matters: a priority-based parallel search strategy generally outperforms sequential revision. Priority search maintains a buffer of candidate sequences, repeatedly selects top-K candidates, proposes K new edits in parallel, evaluates via the structure oracle, and re-inserts all candidates—reducing premature convergence and improving robustness under a strict evaluation budget.
5. Multi-objective optimization is implemented as a weighted sum over ss-pLDDT (confidence), TM-score (global similarity), and Cα-RMSD (geometric deviation), with explicit penalties for large RMSD. Empirically, multi-objective setups improve multiple metrics simultaneously and increase success rates beyond single-objective optimization.
6. The paper emphasizes that residue-level feedback is not sufficient by itself: a compute-matched, information-matched random-mutation baseline (mutating only flagged regions) fails to improve and often degrades fidelity. The gains come from the LLM interpreting feedback and making chemically/structurally informed substitutions.
7. To reduce reward hacking (e.g., poly-alanine runs, motif repetition, length manipulation, or returning the native sequence), RosettaSearch adds soft constraints as textual feedback: repetition checks (6-mer redundancy), explicit length constraints, and (when applicable) ligand-binding position constraints to preserve functional interactions.
8. Generalization checks: improvements persist when final sequences are evaluated by an independent structure predictor (Chai-1), suggesting gains are not merely overfitting to RosettaFold3’s inductive biases. The approach also generalizes across LLM families (o4-mini and Gemini-3), and performance scales with reasoning capability (o3-mini < o4-mini).
9. Beyond native backbones, RosettaSearch also improves designs on de novo Dayhoff atlas backbones (RFDiffusion backbones with ProteinMPNN sequences). Even with strong starting success rates, RosettaSearch increases success-rate-1 from 72.4% to 89.5% and success-rate-2 from 45.5% to 67.3%, notably without providing any reference (native) sequence context.
10. A multi-modal extension uses vision-language models: rendered images of predicted structures are included as feedback to provide spatial context. Performance is comparable to text-only optimization in some settings, while producing qualitatively richer reasoning traces.
📜Paper: arxiv.org/abs/2604.17175
#ProteinDesign #ComputationalBiology #ProteinEngineering #LLM #InferenceTimeSearch #MultiObjectiveOptimization #RosettaFold #StructurePrediction #GenerativeOptimization #MachineLearning
4
6
40
2,303

Apr 23
📢 #highlycited paper
📚 Multiobjective #EnergyConsumption Optimization of a #FlyingWalking #PowerTransmission Line Inspection Robot during Flight Missions Using #ImprovedNSGA_II
🔗 mdpi.com/2076-3417/14/4/1637
👨🔬 by Yanqi Wang et al.
🏫 Shihezi University
#multiobjectiveoptimization

1
3
38
BOAT: Navigating the Sea of In Silico Predictors for Antibody Design via Multi-Objective Bayesian Optimization
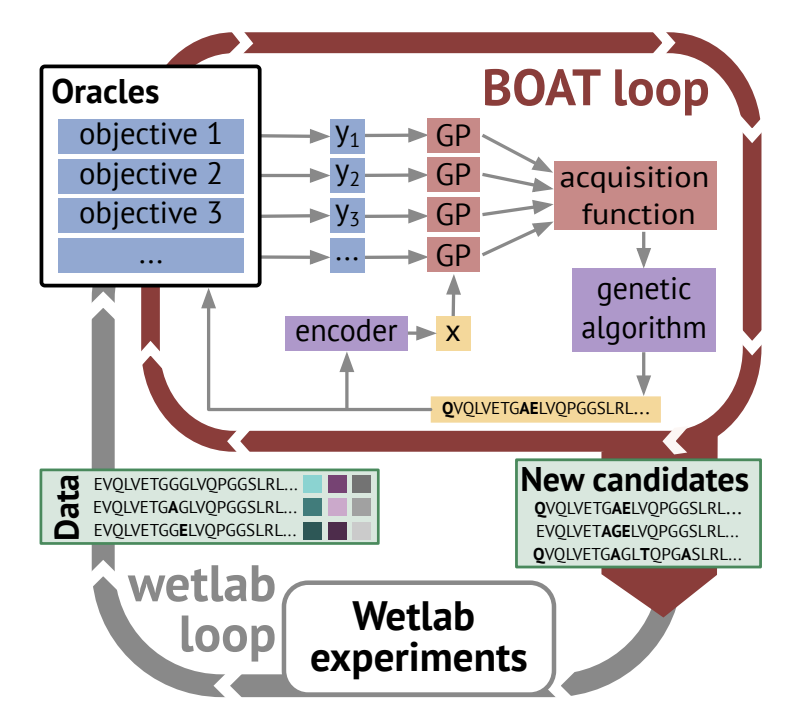
1. BOAT is a plug-and-play multi-objective Bayesian optimization (BO) framework that jointly optimizes multiple antibody properties predicted by arbitrary in silico “oracles,” aiming to replace inefficient sequential filtering pipelines with Pareto-aware design.
2. The key engineering idea: uncertainty-aware surrogate modeling (Gaussian processes) proposes which sequences to score next, while a genetic algorithm (GA) is used to optimize acquisition functions directly in discrete sequence space (avoiding invalid continuous edits and awkward projections).
3. BOAT targets realistic lead-optimization settings where objectives can conflict (e.g., affinity vs. developability vs. immunogenicity risk proxies). It supports full-sequence or region-restricted optimization (e.g., specific CDRs), plus practical constraints such as restricting mutable positions, allowed amino-acid dictionaries, and liability filtering (e.g., glycosylation motifs).
4. Method details: sequences are embedded (one-hot, BLOSUM-derived, bag-of-5-grams, or AbLang-2 embeddings), then modeled with a GP using a Tanimoto kernel to better handle high-dimensional sparse-like representations. Multi-objective acquisition uses EHVI (and NEHVI for noisy settings), implemented via BoTorch.
5. Cross-reactive VHH case study: BOAT optimizes CDR1/2/3 (up to 5 mutations per CDR) to improve binding to two related antigens, optionally adding humanness (OASis) and PLM likelihood (ESM-2) as additional objectives. Mutation choices are constrained to a curated per-position amino-acid dictionary grounded in available experimental single-point data.
6. Benchmarking against GA baselines (sum-of-objectives GA and NSGA-II): across 2–4 objectives and multiple CDRs, BOAT variants reach higher hypervolume earlier and end with better hypervolume under the same oracle-call budget (1000). NSGA-II degrades notably as objective count increases, consistent with many-objective optimization issues.
7. When exhaustive enumeration is feasible (smaller constrained spaces), BOAT recovers Pareto fronts close to the “ground-truth” oracle-induced Pareto frontier, including in very large enumerated CDR3 spaces (tens of millions of sequences), highlighting sample-efficient Pareto exploration rather than brute-force scoring.
8. Diversity matters for wet-lab follow-up: batch BO acquisition (qEHVI/qNEHVI) tends to produce higher Shannon-entropy sequence sets while maintaining strong hypervolume, whereas sequential EHVI can be more exploitative (competitive hypervolume but lower diversity). Larger batch sizes increase diversity, with some early hypervolume trade-offs.
9. Practical limits and regimes: (i) NEHVI can become dramatically slower as objectives increase (e.g., 3 objectives taking minutes per BO step vs seconds for 2), (ii) complex structure-based oracles (Boltz-2 ipTM) can break surrogate fidelity with simple encodings—here, semi-random GA search can be competitive, motivating richer structure-aware surrogates/kernels.
10. Comparison to generative multi-objective methods (LaMBO-2) on the 4-4-20 scFv affinity/expression dataset: using the same discriminative head as BOAT’s oracle, BOAT generally achieves higher hypervolume over generated sequences. However, BOAT can exploit predictor artifacts and go out-of-distribution; adding an ESM-2 likelihood objective acts as a “naturalness” regularizer, underscoring that oracle quality and priors critically shape in silico Pareto fronts.
💻Code: github.com/AstraZeneca/boat
📜Paper: arxiv.org/abs/2604.13980
#BayesianOptimization #MultiObjectiveOptimization #AntibodyDesign #ProteinEngineering #MachineLearning #ComputationalBiology #DrugDiscovery #ActiveLearning #GaussianProcesses #ParetoOptimization
2
15
1,132
Scaffold-Conditioned Preference Triplets for Controllable Molecular Optimization with Large Language Models
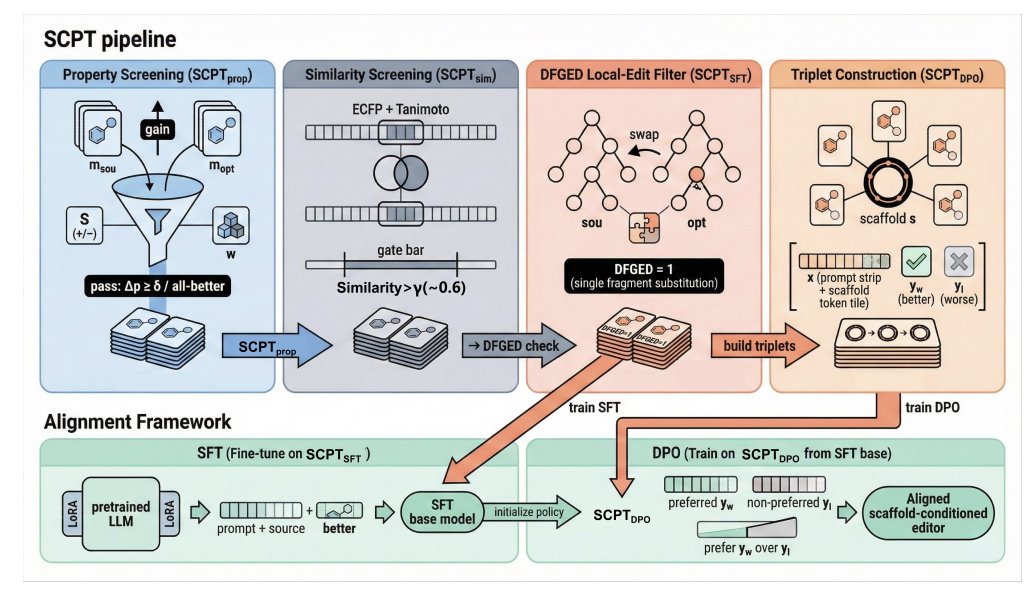
1. The paper frames lead optimization as scaffold-conditioned editing: given a starting molecule, the model must improve one or more properties while staying within a similarity-constrained neighborhood, reducing “high gain but scaffold drift” and “high similarity but negligible gain” failure modes.
2. Core contribution is SCPT (Scaffold-Conditioned Preference Triplets), a reproducible data pipeline that converts static molecular datasets into chemistry-grounded preference supervision of the form <scaffold, better, worse>, explicitly encoding which local edit is preferable under the same scaffold context.
3. SCPT constructs training pairs/triplets via layered chemistry constraints: (i) property-margin filtering (directional gains, single- or multi-objective), (ii) fingerprint Tanimoto similarity thresholding, (iii) enforcing a single-fragment substitution using junction-tree fragment edit distance (DFGEDfrag = 1), and (iv) high atom-level overlap via MCS constraints (near 0.9).
4. The aligned model is a pretrained molecular LLM used as a conditional editor over SMILES/SELFIES. Training is two-stage: supervised fine-tuning (SFT) on scaffold-preserving “worse→better” pairs, followed by Direct Preference Optimization (DPO) on scaffold-anchored preference triplets to shift probability mass toward higher-quality local edits.
5. A key insight is that “data construction knobs” are controllable levers, not incidental preprocessing. By adjusting similarity windows and property-gap thresholds, SCPT yields a predictable similarity–gain frontier: tighter similarity increases SIM but compresses achievable improvement (RI) and can reduce success rate (SR); stricter property gaps trade coverage for sharper preferences.
6. On single-property optimization (8 tasks including DRD2, GSK3β, JNK3, pLogP, QED, BBBP, HIA, Mutag), SCPT-trained editors show near-saturated SR with moderate SIM, while prompted general LLMs often exhibit an unstable trade-off (either high SIM with tiny gains, or higher gains via severe scaffold drift).
7. Multi-objective results highlight the value of scaffold-conditioned preference alignment: in 3- and 4-property tasks (e.g., BDPQ), SCPT DPO substantially improves SR and RI compared to general-purpose and chemistry-specialized LLM baselines, while keeping similarity in a controlled regime rather than unconstrained exploration.
8. Preference alignment is tunable at the model level: sweeping DPO hyperparameters (learning rate, beta) produces consistent shifts—more aggressive updates raise SR/RI but reduce SIM; conservative settings recover similarity but weaken gains—supporting the claim that SCPT provides a usable, controllable preference signal.
9. Against representative non-LLM optimizers (GraphGA, JT-VAE, REINVENT) adapted with similarity-aware objectives, SCPT-trained LLM editors achieve much higher scaffold similarity at matched generation budgets, indicating LLMs are particularly effective for local, scaffold-preserving optimization rather than unrestricted property maximization.
10. The paper also studies data scarcity in higher-order objectives: models trained only on single-property and two-property supervision generalize to unseen three-property combinations with strong SR/RI and substantially higher similarity than non-LLM baselines, suggesting compositional extrapolation from lower-order optimization “primitives.”
📜Paper: arxiv.org/abs/2604.12350
#ComputationalBiology #Cheminformatics #DrugDiscovery #MolecularOptimization #LargeLanguageModels #PreferenceLearning #DPO #ScaffoldHopping #MultiObjectiveOptimization #SMILES
3
18
1,433
Diffusion-based Evolutionary Optimization for 3D Multi-Objective Molecular Generation
1. The paper frames practical 3D molecular design as a Constrained Multi-Objective Optimization Problem (CMOP): optimize conflicting properties while strictly satisfying chemical validity and hard 3D/topological constraints (e.g., multi-fragment assembly), where feasible regions can be disjoint and hard to traverse.
2. Core idea: repurpose a pre-trained SE(3)-equivariant 3D diffusion model as a zero-shot “projection operator” inside an evolutionary algorithm, avoiding expensive retraining for new objectives/constraints while preventing the invalid geometries that standard 3D genetic operators create.
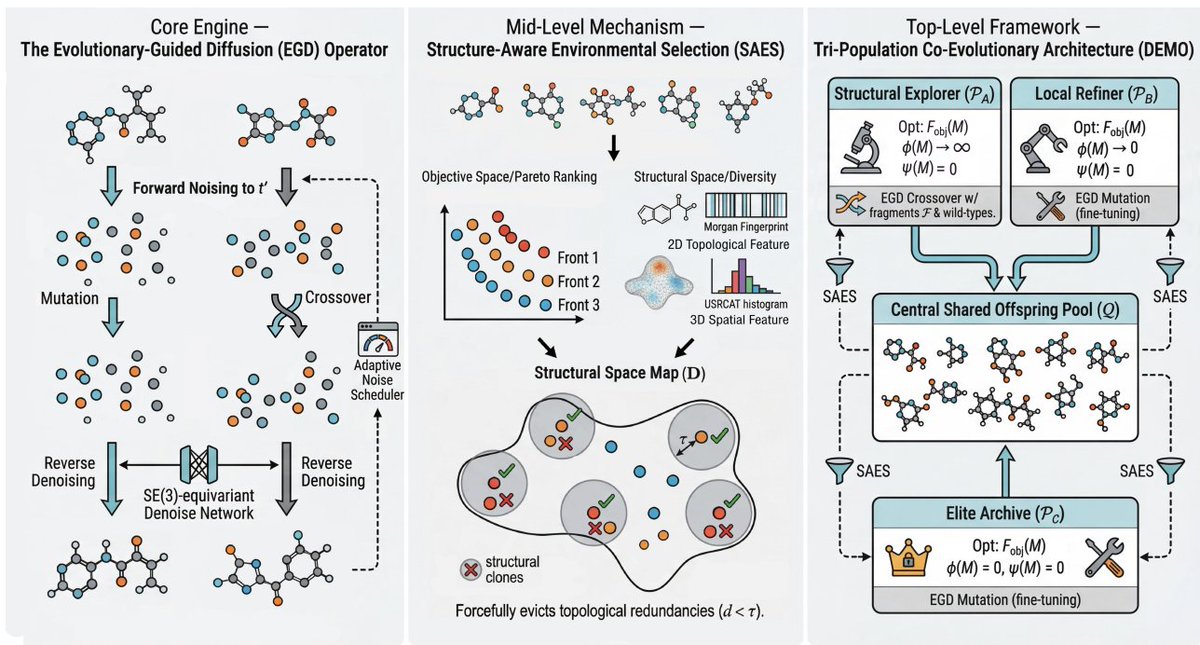
3. Evolutionary-Guided Diffusion (EGD) operator: performs crossover and mutation in diffusion noise space at an optimally chosen intermediate timestep t′, then runs unconstrained reverse diffusion to “heal” chimeric states back onto the valid chemical manifold (resolving valency violations and steric clashes during denoising).
4. Variable-length crossover is handled directly in noise space: EGD samples subset sizes from two parents, concatenates random atomic subsets to form a dimensionally consistent noisy offspring, and relies on reverse diffusion to relax it into a chemically valid molecule—avoiding fixed-length assumptions that break for variable-size molecular sets.
5. Adaptive noise scheduling: the optimal t′ depends strongly on molecular size/complexity. The paper introduces a closed-loop scheduler that uses a coarse binary search followed by Gaussian Process Bayesian optimization to maximize a generative integrity score (Validity Rate × Atom Stability Rate) while penalizing excessive noise.
6. Structure-Aware Environmental Selection (SAES): addresses “loss of structural diversity” in objective-centric MOEAs by enforcing decision-space diversity. It combines 2D topology (Morgan fingerprint Jaccard distance) and 3D conformation/shape (USR atom types descriptor Euclidean distance) into a composite structural distance, then greedily selects candidates with a minimum clearance threshold τ to filter structural clones.
7. DEMO framework (Diffusion-based Evolutionary Molecular Optimization): integrates EGD SAES with a tri-population co-evolution strategy to navigate disjoint feasible regions in CMOPs—PA explores novel scaffolds, PB refines partially feasible intermediates, and PC archives and fine-tunes perfectly feasible elites.
8. Why tri-population matters: ablations show that keeping only fully feasible elites can fail badly in multi-fragment CMOPs because intermediate infeasible structures are necessary stepping stones across disconnected feasible basins; PB specifically maintains and improves these intermediates rather than discarding them.
9. Results summary across tasks (QM9 and GEOM-Drugs): EGD achieves strong zero-shot single- and multi-property targeting (MAE) without retraining; SAES improves Pareto frontier quality while maintaining high uniqueness; DEMO improves CMOP hypervolume while sustaining high Unique Feasible Rate (UFR), and also performs well on 3-objective protein–ligand docking optimization (Vina/QED/SA) with high structural diversity.
📜Paper: arxiv.org/abs/2505.11037
#ComputationalBiology #DrugDiscovery #MolecularGeneration #DiffusionModels #EvolutionaryAlgorithms #MultiObjectiveOptimization #3DGeometry #SE3 #ProteinLigandDocking #FragmentBasedDrugDiscovery
4
17
1,946
Diffusion-based evolutionary optimization for 3D multi-objective molecular generation
1. The paper frames practical 3D molecular design as a constrained multi-objective optimization problem (CMOP): optimize conflicting properties while satisfying strict structural constraints (e.g., multi-fragment assembly), where feasible solutions can be rare and disconnected.
2. It identifies a core mismatch: classical evolutionary operators on raw 3D coordinates break chemical validity (valence, clashes, SE(3) symmetries), while pretrained 3D diffusion models are strong validity-preserving generators but are hard to adapt to new objectives/constraints without retraining.
3. The key operator is Evolutionary-Guided Diffusion (EGD): crossover and mutation are performed in the diffusion model’s continuous noise space (at an intermediate noise level t′), then an SE(3)-equivariant denoiser “projects” the chimeric latent state back onto the valid chemical manifold.
4. EGD supports variable-size “subset crossover” for molecules: it samples atom subsets from two noised parents, concatenates them to form an offspring latent, and relies on reverse diffusion to resolve clashes and restore chemically valid 3D structures; mutation is implemented by noising a parent to t′ and denoising back to yield a meaningful structural perturbation.
5. A central technical detail is choosing the noise level t′: too little noise cannot heal clashes or escape local attractors; too much noise erases inheritance. The paper introduces an Adaptive Noise Scheduler (binary search to find a safe plateau boundary, then Gaussian Process Bayesian optimization) maximizing a composite integrity score (validity rate × atom stability rate) while penalizing excessive noise.
6. To prevent structural mode collapse common in objective-space MOEAs, it proposes Structure-Aware Environmental Selection (SAES), which enforces decision-space diversity using a composite structural distance: 2D Morgan fingerprint Jaccard distance plus 3D USR (with CREDO atom types) distance, then selects with a diversity-first greedy truncation under a minimum-distance threshold.
7. Building on EGD SAES, the full framework DEMO targets CMOPs with a tri-population architecture: a Structural Explorer (PA) to explore diverse valid scaffolds and force fragment assembly, a Local Refiner (PB) to improve partially assembled intermediates, and an Elite Archive (PC) to fine-tune fully feasible molecules—helping traverse disjoint feasible regions without losing stepping-stone intermediates.
8. Experiments span single-property targeting, multi-property targeting, unconstrained MOPs, multi-fragment constrained MOPs, and 3D protein-ligand docking (optimizing Vina score, QED, SA across 10 targets with PoseBusters feasibility checks). DEMO/SAES typically improves hypervolume while maintaining much higher structural uniqueness than objective-centric baselines.
9. Reported takeaways include: (a) EGD can outperform train-free gradient guidance and even retrained conditional diffusion baselines on targeting tasks without retraining; (b) SAES mitigates clone takeover and improves unique-valid/unique-feasible rates; (c) the tri-population design is important—keeping infeasible intermediates materially improves constrained optimization.
📜Paper: arxiv.org/abs/2505.11037
#DiffusionModels #MolecularGeneration #MultiObjectiveOptimization #EvolutionaryAlgorithms #ComputationalChemistry #DrugDiscovery #3DGeometry #ConstrainedOptimization #ProteinLigandDocking #GenerativeModels
1
16
1,766
Symmetric Self-play Online Preference Optimization for Protein Inverse Folding
1. The paper argues that multi-objective inverse folding shouldn’t be forced into a single scalar reward: structural objectives (e.g., self-consistency vs prediction confidence) are only partially aligned, so a single-policy optimizer tends to follow a dominant direction and miss alternative high-quality designs.
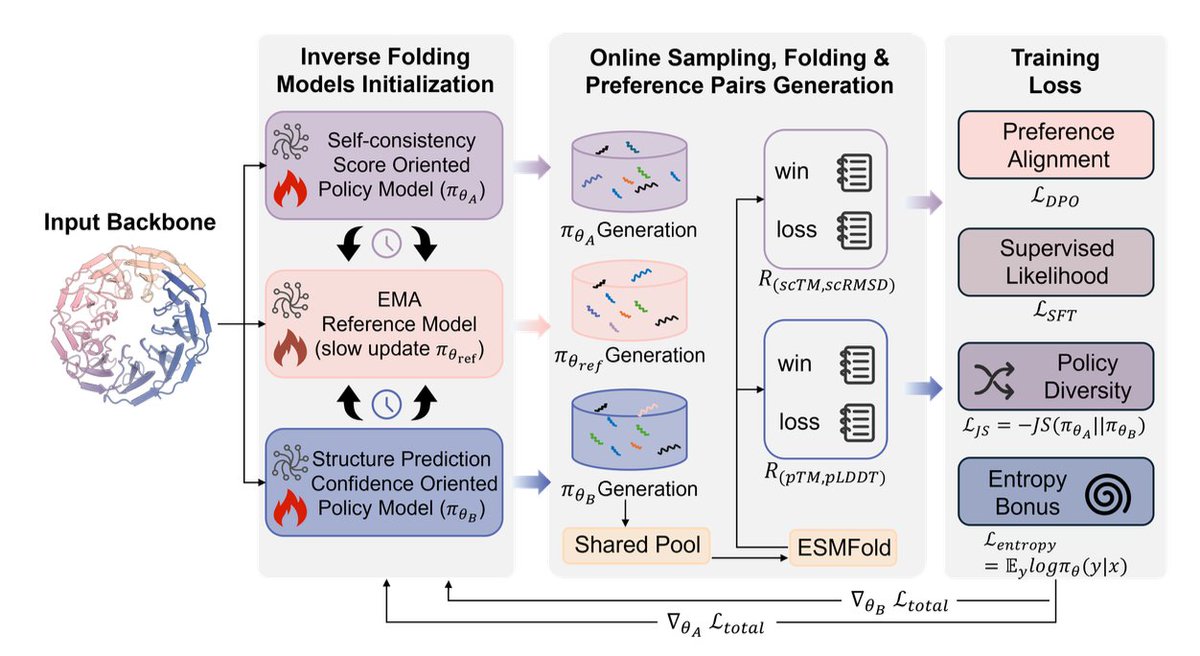
2. It introduces SSP (Symmetric Self-play Preference Optimization), an online preference-optimization framework that keeps two separate policies: one optimized for structural self-consistency (Rsc) and the other for predicted confidence (Rpred), plus an EMA-updated reference model for stabilization.
3. Key mechanism: both policies (and the reference) sample sequences for the same backbone, merge them into a shared candidate pool, refold with ESMFold, filter low-confidence samples, then build preference pairs for each objective from the same pool—creating implicit cross-policy competition and comparison without collapsing objectives.
4. Rewards are explicitly decoupled: Rsc combines scTM and RMSD-based terms after aligning predicted structures to the target backbone; Rpred averages pLDDT and pTM from structure prediction. Training uses DPO-style preference loss plus an SFT term on preferred samples.
5. To prevent policy collapse and encourage complementary exploration, SSP adds (i) a Jensen–Shannon divergence regularizer between the two policies and (ii) an entropy bonus, aiming to cover different regions of the Pareto frontier rather than converging to the same solution.
6. SSP is shown to be architecture-agnostic: implemented on ProteinMPNN (full fine-tuning), ESM-IF1 and ESM3 (LoRA fine-tuning). After training, it produces a single deployable model via merging: task-vector merging for full-parameter models, and weighted LoRA adapter merging for parameter-efficient setups.
7. On native-backbone benchmarks (CATH4.2/4.3), SSP variants consistently improve structure prediction confidence (pTM/pLDDT), self-consistency (scTM, RMSDs), and related metrics over base models and several RL/DPO baselines, indicating that the dual-policy setup improves design self-consistency beyond standard single-policy preference optimization.
8. Generalization is tested on CAMEO43 (targets with max TM-score < 0.5 vs training set). SSP improves pTM/pLDDT and self-consistency vs baselines, supporting the claim that decoupled objectives help in harder, lower-similarity regimes rather than only on in-distribution backbones.
9. Transfer to de novo binder backbones is emphasized as a “real-world” proxy: on BoltzGen-419 (DNA/RNA/peptide binders) and PXDesign-PPI226 (protein binders with specified hotspots), the merged SSP-ESM3 model leads across pTM/scTM and interface confidence (ipTM), and achieves strong design success rates—suggesting robustness across binder types and backbone generators.
10. The paper provides interpretability evidence that the two objectives truly induce different optimization directions: white-box analysis of ESM3 LoRA updates shows low subspace overlap (SVD-based) and near-orthogonal update directions (cosine similarity near 0) across many layers, with only mild alignment in deeper layers—supporting the “partially aligned objectives” hypothesis.
💻Code: github.com/wwzll123/SSP
📜Paper: biorxiv.org/content/10.64898…
#ProteinDesign #InverseFolding #ReinforcementLearning #PreferenceOptimization #MultiObjectiveOptimization #ComputationalBiology #ESM3 #ProteinMPNN #LoRA #SelfPlay
1
20
1,522
How to Make the Most of Your Masked Language Model for Protein Engineering
1 The paper introduces a new sampling method for masked language models (MLMs) in protein engineering that significantly outperforms existing approaches, with the key finding that choice of sampling algorithm matters at least as much as choice of model.
2 The core innovation is temperature-annealed stochastic beam search, which reframes generation as sequence evaluation rather than mutation-centric sampling, exploiting MLMs' efficiency at evaluating pseudoperplexity of entire 1-edit neighborhoods.
3 This sequence-centric approach enables flexible multi-objective optimization with black-box scoring functions, overcoming limitations of mutation-centric methods that struggle with non-differentiable scores like OASis percentile or isoelectric point.
4 The method achieves dramatic computational speedups: O(BEL³) complexity compared to O(EL³) per sequence for traditional approaches, yielding 20EL× speedup for realistic scenarios while producing higher-quality sequences.
5 Extensive in vitro experiments on actual antibody therapeutics campaigns reveal that ESM-2 (trained on generic proteins) and AbLang-2 perform exceptionally well, with beam search consistently outperforming Gibbs sampling across all models.
6 When combined with supervised guidance using smooth Tchebycheff scalarization, the approach achieved 100% success rate in synthesizability and binding QC criteria, while Pareto-based non-dominated sorting showed competitive but slightly lower performance.
7 The work provides practical recommendations: use supervision when possible, prefer AbLang-2 or ESM-2-650M for antibody engineering, adopt stochastic beam search over Gibbs sampling, and carefully consider scalarization methods for multi-objective guidance to avoid undesirable side effects.
📜Paper: arxiv.org/abs/2603.10302
#proteinengineering #antibodydesign #machinelearning #bioinformatics #computationalbiology #drugdiscovery #languagemodels #MLM #beamsearch #multiobjectiveoptimization
7
33
2,972
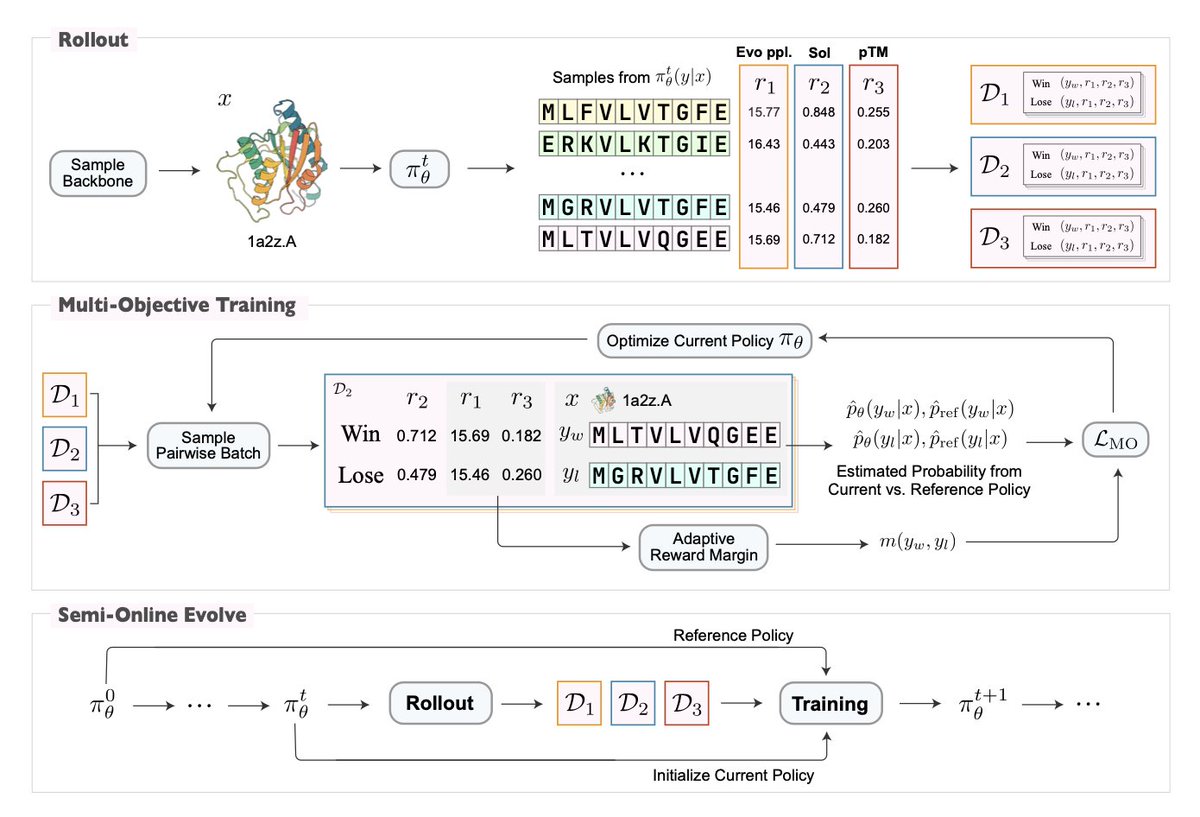
Property-Driven Protein Inverse Folding with Multi-Objective Preference Alignment
1 Researchers introduce ProtAlign, a multi-objective preference alignment framework that fine-tunes pretrained inverse folding models to optimize for multiple developability properties simultaneously without sacrificing structural fidelity.
2 The core innovation lies in a semi-online Direct Preference Optimization (DPO) strategy with an adaptive preference margin that automatically resolves conflicts between competing objectives like solubility and thermostability.
3 Unlike existing approaches that rely on post-hoc mutation, inference-time biasing, or retraining on curated subsets, ProtAlign enables target-independent optimization that requires minimal domain expertise and hyperparameter tuning.
4 Applied to ProteinMPNN, the resulting model MoMPNN achieves superior performance on solubility and thermostability benchmarks compared to specialized models like SolubleMPNN and HyperMPNN, while maintaining or improving designability metrics.
5 The framework constructs preference pairs using in silico property predictors and employs a flexible margin mechanism that reduces the required preference gap when winning sequences perform worse on auxiliary properties, preventing over-optimization of single objectives.
6 MoMPNN demonstrates robust generalization across diverse evaluation scenarios including CATH 4.3 crystal structures, de novo backbones generated by RFDiffusion, and real-world binder design tasks, outperforming baselines consistently.
7 The semi-online training paradigm decouples rollout and evaluation from training, enabling efficient batch computation and avoiding the computational overhead of running property predictors during gradient updates.
💻Code: github.com/biogeometry/ProtA…
📜Paper: arxiv.org/abs/2603.06748
#ProteinDesign #InverseFolding #MultiObjectiveOptimization #DirectPreferenceOptimization #ComputationalBiology #ProteinMPNN #MachineLearning #StructuralBiology
5
37
2,672

Feb 26
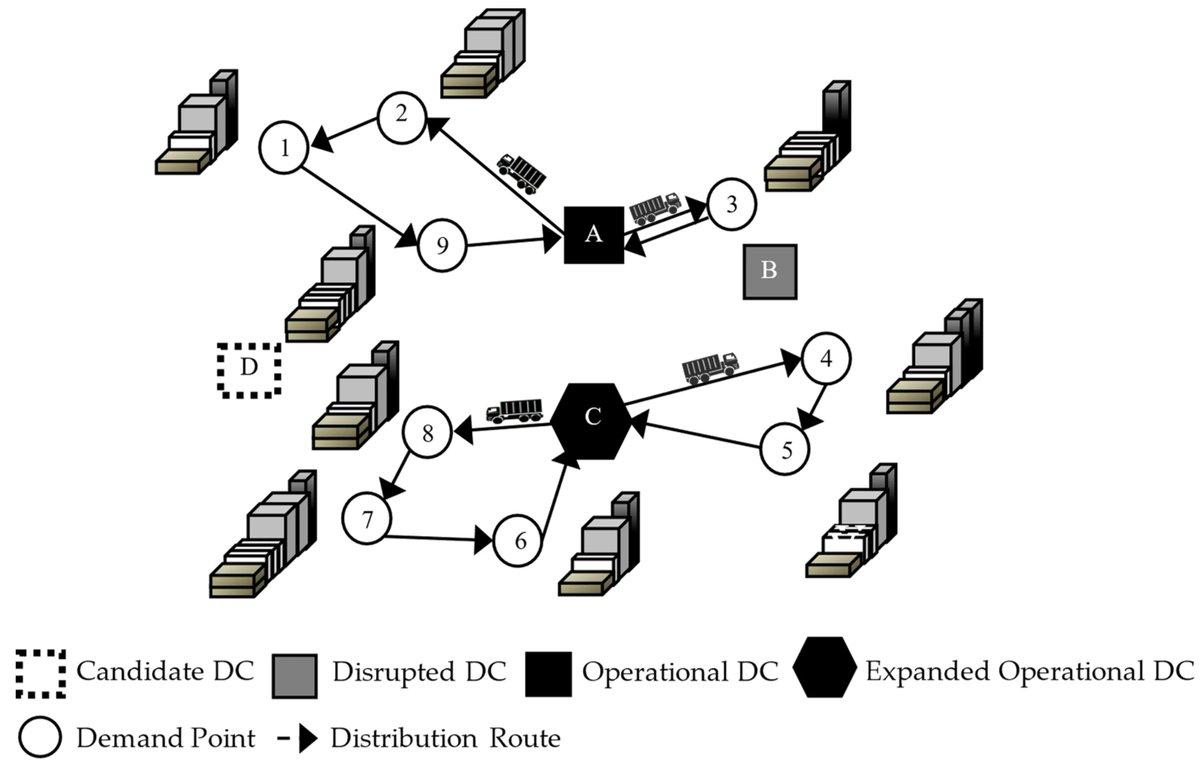
Post-Earthquake #EmergencyLogistics Location-Routing Optimization Considering Vehicle Three-Dimensional Loading Constraints
✏️ Xujin Pu and Xu Zhao
🔗 brnw.ch/21x0gzg
Viewed: 3214; Cited: 10
#mdpisymmetry #multiobjectiveoptimization
1
2
68
Multi-objective optimization and quantum hybridization of equivariant deep learning interatomic potentials on organic and inorganic compounds
1 A research team from Terra Quantum AG and collaborators has developed enhanced variants of the Allegro machine learning interatomic potential (MLIP) model, achieving superior accuracy through multi-objective hyperparameter optimization and novel quantum-classical hybrid architectures.
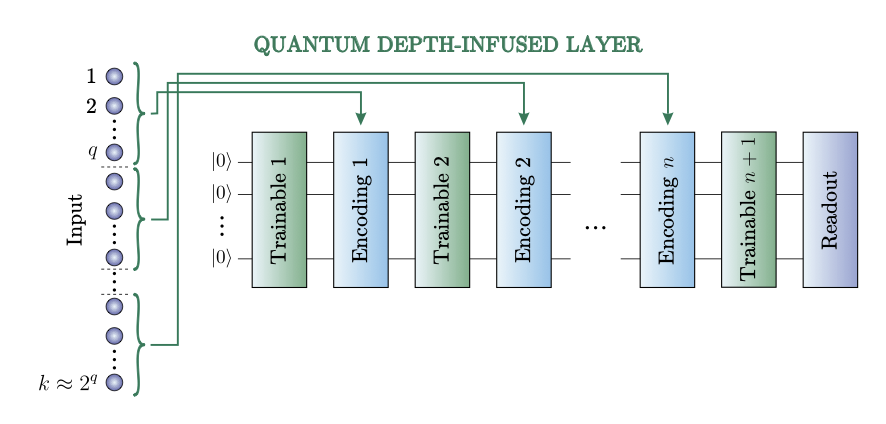
2 The study introduces two key innovations: an extended Allegro model with additional classical MLP layers (Allegro MLP) and a quantum depth-infused variant (Allegro QDI) that incorporates variational quantum circuits into the neural network architecture.
3 Using their implementation of the SAMO-COBRA multi-objective optimization algorithm, the researchers simultaneously optimized for prediction accuracy (force/energy MAE) and inference speed across four diverse datasets: QM9 organic molecules, rMD17 aspirin and benzene subsets, and a newly generated copper-lithium inorganic dataset.
4 The Allegro QDI model demonstrated remarkable performance on the Cu-Li dataset, achieving a 13% improvement in force prediction accuracy over the baseline Allegro model, showcasing the potential of quantum-enhanced architectures for materials science applications.
5 The quantum layer employs a data re-uploading strategy that sequentially encodes features onto a limited qubit lattice with entangling gates, enabling complex nonlinear pattern recognition without requiring one qubit per feature.
6 Comprehensive quantum circuit analysis using ZX calculus revealed 100% parameter preservation after simplification, Fisher information metrics confirmed good trainability without barren plateaus, and Fourier analysis showed 79% expressivity with 127 non-zero coefficients out of 161 possible terms.
7 The Cu-Li dataset generation represents a significant contribution itself: 11,635 DFT-calculated structures including surface vacancies, adatoms, coherent interfaces, and melt-quench amorphized interfacial structures for modeling high-temperature Li-Cu material behavior.
8 Pareto front analysis revealed dataset-dependent trade-offs between accuracy and inference time, with Allegro MLP generally dominating on organic molecules while Allegro QDI showed particular strength on the inorganic metallic system.
💻Code: github.com/glatq/allegro
📜Paper: arxiv.org/abs/2602.16908
#MachineLearning #QuantumComputing #MaterialsScience #InteratomicPotentials #EquivariantNeuralNetworks #MultiObjectiveOptimization #DFT #MolecularDynamics #ComputationalChemistry #QuantumMachineLearning
5
1
9
1,246
Jan 28
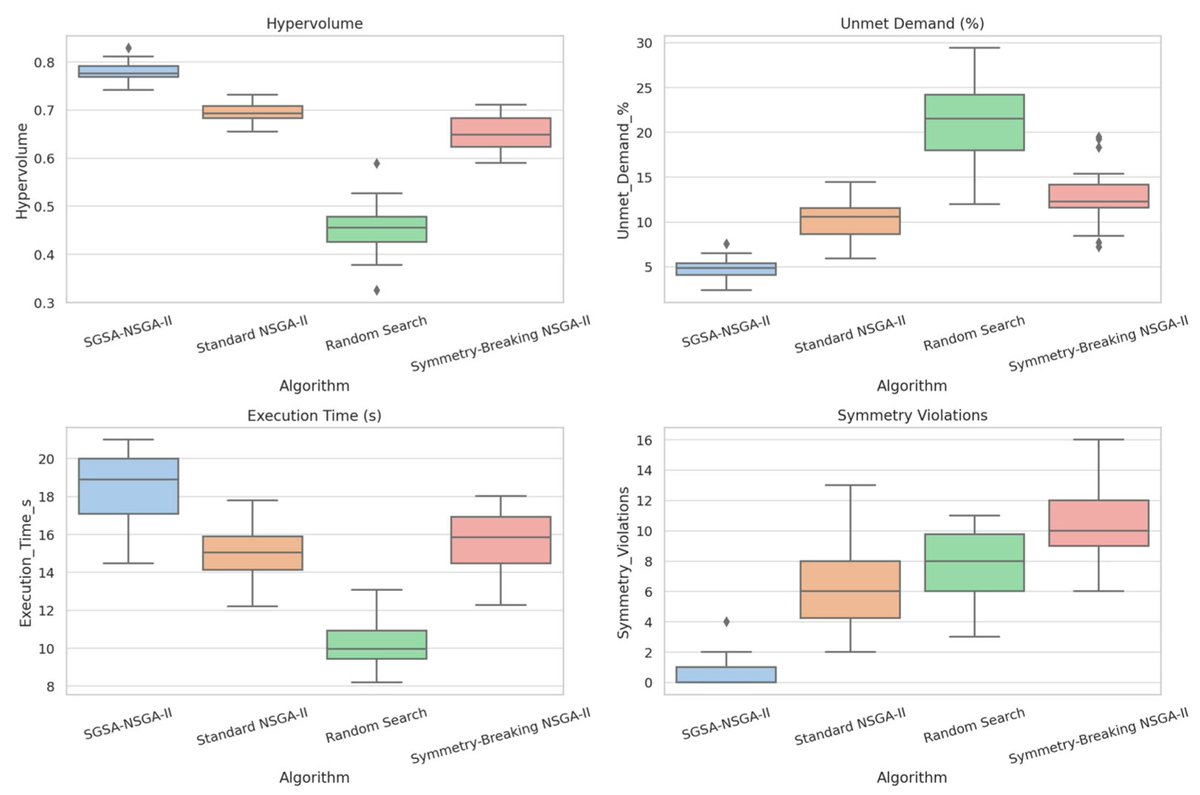
Check this newly published article "Symmetry-Guided Surrogate-Assisted NSGA-II for #MultiObjectiveOptimization of #RenewableEnergySystems" at brnw.ch/21wZrFv
Author: Manuel J. C. S. Reis
#mdpisymmetry #metaheuristics #surrogatemodeling
@UTAD_oficial
@ComSciMath_Mdpi
2
3
46

Jan 16
Just released version 9.0.0 of the #Jenetics #Java library. It now requires Java 25 to compile and run.
#GeneticAlgorithm #EvolutionaryAlgorithm #GrammaticalEvolution #GeneticProgramming #MultiobjectiveOptimization #MOEA
github.com/jenetics/jenetics…
1
2
161

22 Dec 2025
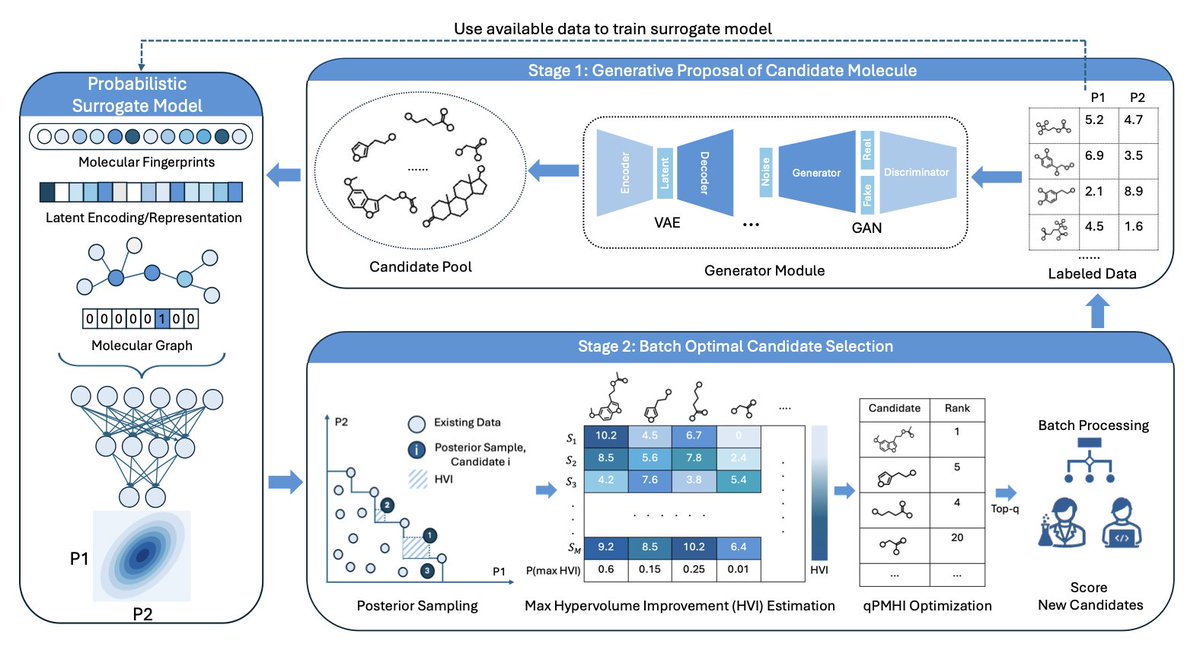
Generative Multi-Objective Bayesian Optimization with Scalable Batch Evaluations for Sample-Efficient De Novo Molecular Design
1. A novel “generate-then-optimize” framework is introduced for de novo molecular design, which decouples candidate generation from selection, allowing for more scalable and efficient multi-objective optimization in chemical space.
2. The framework proposes a new acquisition function, qPMHI, which enables exact and scalable batch selection by ranking candidates based on their probability of maximum hypervolume improvement. This avoids complex combinatorial optimization.
3. The method is validated on both synthetic benchmarks and a real-world application in sustainable energy storage, demonstrating significant improvements in identifying high-performing molecules with fewer queries compared to existing methods.
4. The modular design allows for flexibility in choosing generative models and surrogate property predictors, making it adaptable to various molecular representations and design tasks. This reduces architectural entanglement and improves robustness.
5. The study highlights the potential of this approach for accelerating molecular discovery in large, open-ended chemical spaces, especially when leveraging parallel experimental platforms or high-throughput simulations.
📜Paper: arxiv.org/abs/2512.17659v1
#BayesianOptimization #MolecularDesign #GenerativeModels #MultiObjectiveOptimization #Chemistry #AI
1
2
27
1,983