
Jun 13
The next billion-dollar biotech company won't be called BioTechSolutionsAI.
It will be called something people remember.
PYP.ai
Predict Your Protein.
Pick Your Peptides.
A rare 3-letter .AI brand for the future of protein intelligence, longevity, precision medicine, and AI-driven health.
Available for acquisition
#AI #Biotech #Longevity #HealthTech #PrecisionMedicine #ProteinEngineering #Peptides #Startup

1
4
100

Sequence-based therapeutic peptide classification with augmented negative sampling
1 The paper introduces TheraPep-AI, a sequence-only multi-label classifier covering 48 therapeutic functions (plus a global “is_therapeutic” flag) trained on 54,655 natural peptides from TheraPepDB, aiming to make peptide screening more practical by explicitly controlling false positives.
2 The central technical idea is augmented negative sampling: instead of treating “non-therapeutic” as an afterthought, the authors generate diverse synthetic decoy peptides that closely match therapeutic peptides’ composition statistics but are unlikely to contain functional motifs, forcing the model to learn discriminative sequence order signals rather than shortcuts.
3 Decoys are generated with a four-tier difficulty ladder using Markov-style statistics: uniform random (easy), global first-order Markov (matches dipeptides), position-dependent Markov (matches positional biases), and class-frequency sampling (matches per-class amino acid composition). Decoy lengths are sampled from the therapeutic length distribution to prevent length-based cheating. Training uses an overall 1:2 positive:negative ratio with equal contributions from the four decoy types.
4 On the controlled decoy benchmark, prior models show very high false positive behavior (reported as 60–70% on these peptide-like negatives), while TheraPep-AI reduces FPR dramatically; the final model reports 2.1% FPR on held-out synthetic negatives, highlighting that negative design is a first-class component of evaluation, not just training.
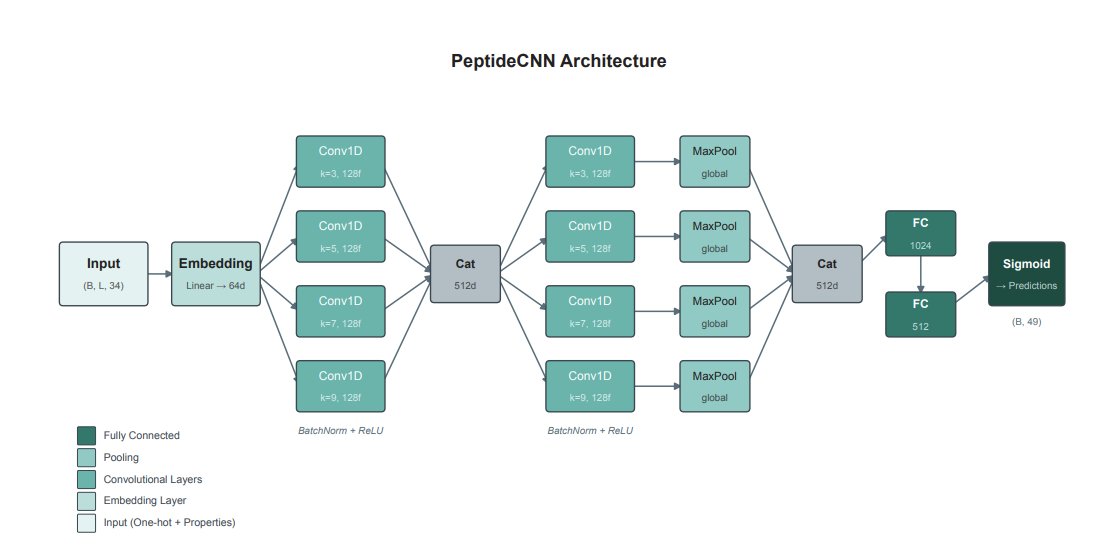
5 The modeling choice is deliberately compact: a two-layer multi-scale CNN with parallel kernel sizes {3,5,7,9} (motif-scale windows), global max pooling for position invariance, and a 34D per-residue encoding (one-hot physicochemical properties). A lightweight ~1M parameter variant targets fast screening; a fine-tuned 5-model ensemble totals ~15M parameters.
6 On TheraPepDB test positives, the fine-tuned ensemble achieves 79.9% Micro F1 and 54.6% Macro F1 across 48 functions, reflecting strong performance on abundant classes while still improving coverage for rarer categories via class-weighted BCE (square-root weighting with caps explored).
7 Head-to-head retraining on identical TheraPepDB splits shows the CNN’s practical advantage: TPpred-LE (transformer encoder-decoder) reaches similar positive-set Micro F1 but has much higher FPR on the synthetic negatives (13.9% vs 2.1%), while PrMFTP achieves very low FPR (1.0%) but substantially worse positive-set F1—underscoring the precision/recall/FPR trade space when negatives are made realistic.
8 External generalization is tested on the TPpred-LE benchmark (12 shared labels, 1,024 sequences) with zero sequence overlap enforced by removing shared sequences from training. TheraPep-AI trained on TheraPepDB reaches 55.3% Micro F1 and 38.6% Macro F1 on the 12 labels, close to TPpred-LE’s in-domain baseline (57.9%/38.1%), while a TPpred-LE architecture retrained on TheraPepDB drops sharply (18.8% Micro F1), suggesting motif-centric, position-invariant CNN features transfer better across datasets.
9 Interpretability is addressed via an L1-regularized sparse variant (85–98% sparsity in conv layers) that enables filter-level analysis. Filters correlate with labels and recover recognizable motif detectors (e.g., GTFT/GTFTS for glucagon-like metabolic peptides, cysteine-rich patterns for defensin-like antimicrobials, and specific charged motifs for AntiHIV), providing evidence the network learns biologically meaningful sequence patterns and also learns “non-therapeutic” signals (statistical improbabilities) useful for rejection.
💻Code: github.com/terra-quantum-pub…
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #MachineLearning #DeepLearning #Peptides #DrugDiscovery #ProteinEngineering #CNN #MultiLabelClassification #Preprint
9
1,065
DeepRHP: A Hybrid Variational Autoencoder for Designing Random Heteropolymers as Protein Mimics
1. The paper introduces DeepRHP, a semi-supervised hybrid VAE that learns latent representations of random heteropolymer (RHP) sequence ensembles while explicitly constraining the latent space to reflect function-related chemical features, aiming to make RHP design more data-driven than empirical screening.
2. Key architectural idea: a classical sequence VAE is paired with a parallel feature-based VAE that reconstructs a deterministic chemical feature y derived from the same sequence x; both branches share the same latent variable z, encouraging z to encode both sequence-pattern statistics and chemically meaningful structure.
3. The training objective modifies the standard VAE ELBO by combining two reconstruction terms: (a) discrete sequence reconstruction (cross-entropy over monomer tokens) and (b) feature reconstruction (MSE on y), weighted by a tunable α, while keeping the KL regularization on q(z|x) vs p(z).
4. The “feature” used for semi-supervision is the sliding-window average hydrophilic–lipophilic balance (HLB), motivated by prior evidence that local hydrophobicity/solubility patterning is strongly tied to RHP behavior in protein stabilization and transport applications.
5. Data pipeline: the study simulates 10,000 RHP sequences per monomer composition using Compositional Drift (copolymer models Monte Carlo), focusing on a 4-methacrylate monomer set (MMA, EHMA, OEGMA, SPMA) spanning hydrophobic, very hydrophobic, hydrophilic, and charged chemistries.
6. To connect synthetic polymers to biology, ~30k membrane and ~30k globular protein sequences (UniProt, 50% identity threshold) are reduced into a 4-letter “monomer-equivalent” alphabet based on residue hydrophobicity/charge, enabling joint embedding and similarity analysis between proteins and RHP ensembles.
7. Design insight 1 (alphabet size): by comparing 2-monomer vs 4-monomer RHP libraries in the learned latent space (visualized via PCA of latent factors), the paper argues that 2-monomer sequence space is too broad relative to protein-like regions, whereas 4-monomer libraries yield more localized, protein-overlapping distributions—supporting why four monomers can be “enough” for protein-mimic behavior.
8. Design insight 2 (composition): within a fixed 70% hydrophobic / 30% hydrophilic constraint, varying the MMA:EHMA ratio produces distinct RHP ensembles; DeepRHP’s latent-space overlap with Aquaporin Z (AqpZ) projections highlights specific compositions (notably matching the published optimal formulation) as most similar to the target membrane protein.
9. Practical takeaway: DeepRHP reframes RHP design as an ensemble-level representation learning problem—enabling composition suggestion by latent-space similarity to target proteins—without requiring exact polymer sequences, 3D structures, or multiple sequence alignment, and with a plug-in pathway to incorporate other chemical features beyond HLB.
10. The authors report ablations indicating the hybrid (feature-guided) architecture outperforms a classical VAE alone for producing useful latent structure, while noting that current evaluation is largely qualitative and motivating future quantitative metrics and downstream tasks (e.g., membrane protein subclass discrimination, RHP–protein similarity scoring).
📜Paper: arxiv.org/abs/2606.11651
#ComputationalBiology #MachineLearning #DeepLearning #VAE #GenerativeModels #PolymerScience #MaterialsInformatics #ProteinEngineering #MembraneProteins #Cheminformatics

2
14
1,165

Jun 12
MKUltra never ended—it evolved into radio-genetics and remote neural monitoring. This isn't a conspiracy theory. It's a conspiracy fact. This is a black-budget reality. This is the single most dangerous, state-sanctioned conspiracy in human history and several intelligence agencies around the world are using it on its citizens #targetedindividuals #radiogenetics #proteinengineering #computerbraininterface #anomoloushealthincidents #havanasyndrome

1
1
33

🧪🦠 A faster way to produce and protect industrial enzymes!
📖Read more at: mdpi.com/2674-0583/3/1/5
#SyntheticBiology #CellFreeProteinSynthesis #Biocatalysis #Enzymes #Biotechnology #VirusLikeParticles #ProteinEngineering #IndustrialBiotechnology #Nanobiotechnology
9
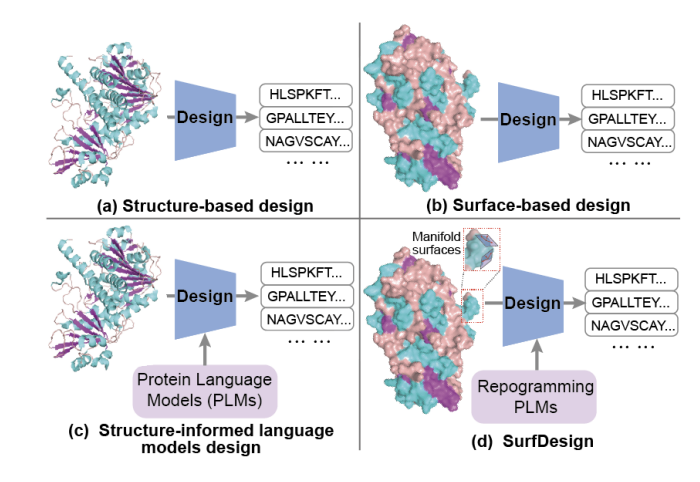
SurfDesign: Effective Protein Design on Molecular Surfaces
1. SurfDesign reframes protein design around molecular surfaces (shape physicochemical complementarity), aiming to better control functional regions like binding interfaces and enzyme pockets compared with backbone-only conditioning.
2. Core technical idea: treat molecular surfaces as continuous geometric manifolds rather than unordered point clouds/meshes, so the model can leverage local tangent structure, curvature, and directional consistency that are important for interaction-centric design.
3. The method builds an oriented surface point cloud Q where each point includes coordinates, a unit normal vector, and physicochemical attributes (e.g., hydrophobicity, charge, H-bond features). Surfaces are generated via PyMOL/MSMS and denoised with Gaussian smoothing; no residue identities, MSAs, or functional labels are used in surface generation.
4. SurfDesign introduces a Surface-conditioned Equivariant Message Passing (SEMP) encoder: SE(3)-equivariant updates use invariant radial distances, curvature descriptors (from local covariance eigenvalues), and directional angular features derived from surface normals (two intersection angles one dihedral angle).
5. Directionality is encoded with spherical Fourier–Bessel bases over distances and angles; messages are attention-reweighted and used to update both node features and coordinates, with optional per-layer recomputation of normals/curvatures to stay consistent as coordinates evolve.
6. To address limited surface-structure paired data and improve sequence priors, SurfDesign integrates pretrained protein language models (PLMs) via parameter-efficient fine-tuning (hybrid PEFT: structural adapter LoRA), trained with conditional masked language modeling rather than autoregressive decoding.
7. Binder design benchmark (6 targets) uses AF2 pAE_interaction as a functional proxy. SurfDesign achieves the best average pAE_interaction (15.85) and the highest overall success rate (30.14%), outperforming SurfPro (surface-conditioned baseline) and backbone-only baselines like ProteinMPNN, PiFold, and LM-DESIGN.
8. Enzyme design benchmark (5 enzyme–substrate systems; leakage-controlled by excluding overlaps with CATH pretraining) uses ESP score as a proxy for enzyme–substrate compatibility. SurfDesign attains the best average success rate (47.30%) and the best average ESP under greedy decoding (0.9058), with gains persisting in a zero-shot substrate setting.
9. Inverse folding is positioned as a diagnostic for structural compatibility (not “recovering the native sequence”). SurfDesign reports strong results on CATH splits, including perplexity 2.41 and AAR 74.13% on CATH 4.2, plus improved surface recovery metrics (IoU/CD/NC) versus PLM-based baselines; scaling larger PLMs further improves recovery.
💻Code: github.com/smiles724/SurfDes…
📜Paper: arxiv.org/abs/2606.07567
#ProteinDesign #ComputationalBiology #GeometricDeepLearning #ProteinEngineering #EnzymeDesign #ProteinBinding #EquivariantNetworks #ProteinLanguageModels #KDD2026
15
74
3,952
Discriminator-guided Inverse Folding for Multi-property Protein Design
1 DGIF introduces a plug-and-play way to steer an inverse-folding model toward multiple protein properties at once, without fine-tuning the generative model and without needing multi-property-labeled datasets.
2 Core idea: during autoregressive sequence generation, DGIF backpropagates gradients from an auxiliary discriminator into the decoder’s internal history state (KV-cache / “history states”), then re-samples the next residue from the updated distribution—repeating this at every step.
3 The discriminator is a composition of multiple single-property predictors. Each predictor can be trained independently on a dataset labeled for only that property, and DGIF combines their signals with weights (beta_i) to perform multi-objective optimization.
4 DGIF is implemented on top of ESM-IF1, producing three variants: DG-Thermo (thermostability), DG-Sol (solubility), and DG-Dual (thermostability solubility). The base inverse-folding model parameters remain unchanged.
5 For thermostability guidance, the paper trains a ΔΔG predictor using ESM-IF1 representations on the Megascale dataset (≈700k mutation–stability pairs), with additional evaluation on FireProt and S669. The predictor outperforms several classic baselines (e.g., FoldX/Rosetta/Thermonet) and is competitive with ThermoMPNN.
6 DG-Thermo improves design outcomes vs unguided ESM-IF1 on: (i) average top-K recall for stabilizing mutations on Megascale test proteins, and (ii) “success rate” of full-sequence designs that both improve predicted stability (ΔΔG > 1.0 kcal/mol) and maintain foldability (predicted structure RMSD < 2 Å).
7 Mechanistic signals emerge naturally: DG-Thermo-designed proteins show more salt bridges and hydrophobic interactions, and amino-acid composition shifts consistent with thermophilic trends (e.g., increased L/P/R/W and decreased D/K/M/Q), despite these rules not being explicitly encoded as constraints.
8 MD validation: for xylanase at 450 K (100 ns), DG-Thermo variants maintain structure (lower RMSD, higher secondary-structure retention) compared with wild type and an unguided ESM-IF1 design; additional CATH-sampled scaffolds show similar stability gains in MD.
9 Solubility guidance: a binary solubility predictor (ESM-IF1 representations MLP) is trained on Khurana et al. and tested on Chang et al.; DG-Sol improves top-K recall on SoluProtMutDB and increases design success rates under joint criteria (better predicted solubility RMSD < 2 Å). On membrane proteins, DG-Sol designs increase surface polar residue proportion, consistent with higher solubility.
10 Multi-property optimization: DG-Dual jointly applies thermostability and solubility predictors and shifts designs toward the Pareto front (better stability/solubility trade-offs) on CATH redesign tasks. Wet-lab validation on Rhodococcus ruber alcohol dehydrogenase (RrADH) tests 10 DG-Dual-suggested single mutations: all improve solubility; 8/10 increase melting temperature. Examples include A50E (≈2x ELISA solubility signal 2.79 °C Tm) and S223A ( 6.47 °C Tm with concurrent solubility gain).
💻Code: github.com/aweqardf/ESM-IF1-…
📜Paper: doi.org/10.1002/advs.75988
#ProteinDesign #InverseFolding #MultiObjectiveOptimization #ESM #ComputationalBiology #MachineLearning #Thermostability #Solubility #ProteinEngineering

5
23
1,446
Flexible Kernels for Protein Property Prediction
1. The paper introduces LOCK-GP: Gaussian processes with a new protein sequence kernel that combines evolutionary substitution matrices (e.g., BLOSUM) with an explicit “local linearity” inductive bias to model protein property landscapes from sparse experimental data.
2. Key kernel idea (LOCK: Locally Linear Correlation Kernel): replace one-hot “same/different” comparisons with amino-acid similarity from substitution matrices, and learn landscape-specific Hadamard-power exponents to tune how strongly similarities are amplified/attenuated while preserving kernel validity.
3. A central technical observation: many BLOSUM matrices are not only PSD but also infinitely divisible, so elementwise exponentiation by any positive power preserves PSD. This enables learnable exponents inside the GP kernel without breaking positive semidefiniteness.
4. LOCK is built from (i) an additive “linear” correlation kernel and (ii) a multiplicative “RBF-like” correlation kernel, then combined so predictions are nuanced and non-linear near training data but revert to a robust linear predictor farther away (avoiding both aggressive linear extrapolation and mean-reversion to the prior mean).
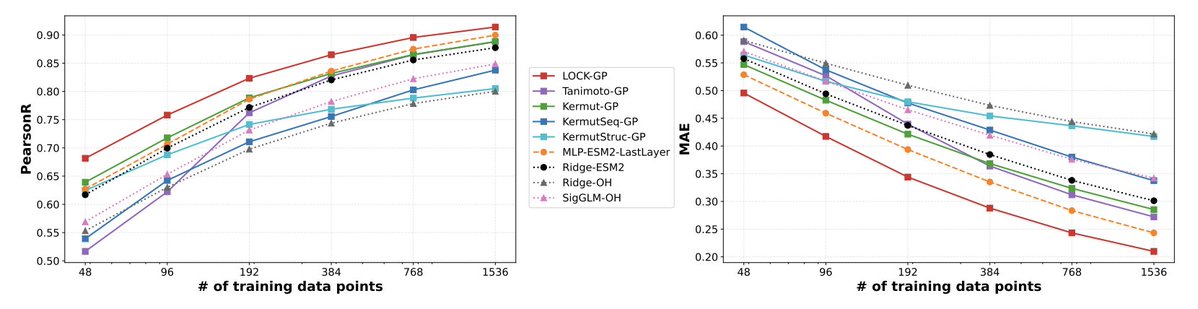
5. Benchmarking is extensive: 30 predictors evaluated across 21 protein property datasets (thermostability, binding affinity, fluorescence, capsid viability, etc.) under three regimes: i.i.d. CV, Hamming-distance extrapolation, and an “unseen mutations” OOD regime where test sequences include mutations absent from training.
6. Results highlight data efficiency and uncertainty quality: across datasets and training sizes (e.g., 48–1536 points), LOCK-GP is typically best or near-best on correlation and error metrics, and shows strong calibration via proper scoring rules like CRPS; uncertainty improves notably when local linearity is included.
7. A notable empirical takeaway: a sequence-only LOCK-GP that relies on a small substitution-matrix prior can frequently outperform or match baselines that depend on large foundation models (e.g., ESM-2 embeddings, structure features, ProteinMPNN-derived features), especially in extrapolation and OOD “unseen mutation” settings where high-dimensional embeddings can be fragile.
8. The paper generalizes LOCK to CLOCK (structure-conditioned LOCK): positional structure embeddings from a foundation model are mapped to position-specific amino-acid correlation matrices (parameterized as exp(-||z_a - z_a'||^2)), effectively learning structure-aware substitution behavior that can be used “zero-shot” as a kernel prior and then refined by GP training.
9. Multi-task learning: CLOCK-GP is trained across 371 thermostability landscapes (Tsuboyama et al.), showing that learning a shared, structure-conditioned kernel across landscapes yields strong performance; CLOCK-GP is especially competitive in low-landscape regimes (e.g., training on 10 landscapes), and learned correlations are interpretable (e.g., proline preferences near helix N-termini; arginine favored on surfaces vs cores).
10. Additional demonstrations: LOCK-GP supports GP-based Bayesian optimization via Thompson sampling to control exploration/diversity in design, and extends to binary classification (e.g., quantized fluorescence) with strong accuracy scaling with dataset size.
💻Code: github.com/generatebio/lock_…
📜Paper: arxiv.org/abs/2606.11057
#ComputationalBiology #ProteinEngineering #GaussianProcesses #MachineLearning #Kernels #ProteinDesign #UncertaintyQuantification #MultiTaskLearning #FoundationModels #Bioinformatics
2
11
1,126
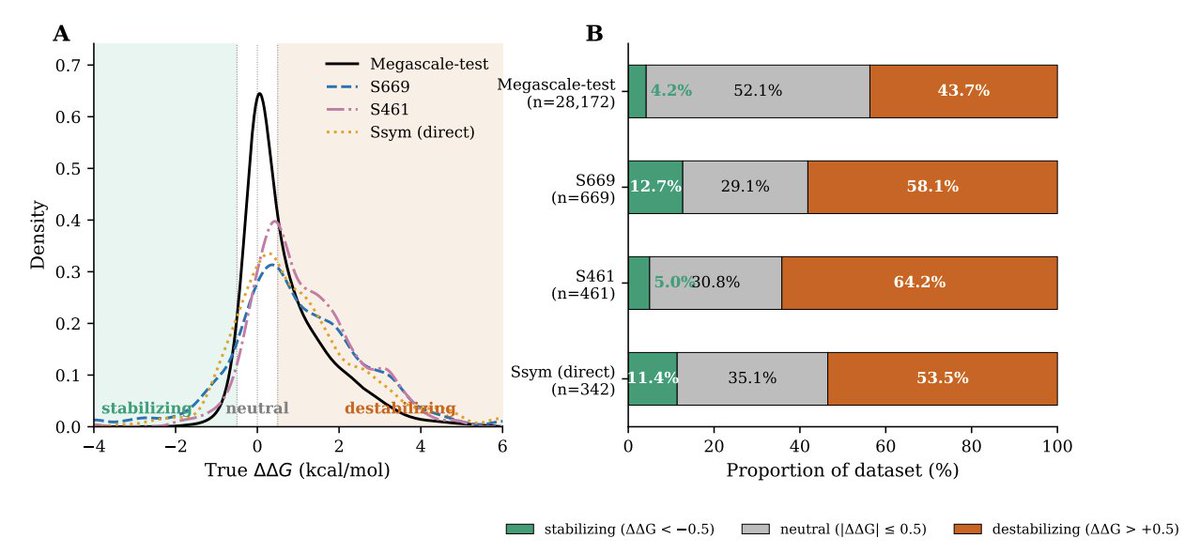
Constraint-Aware Optimization for Robust Protein Stability Prediction
1. The paper proposes an optimization-level framework (no architecture changes) to improve robustness of multimodal protein stability (ΔΔG) predictors built on the SPURS-style backbone (ESM2 sequence ProteinMPNN structure, fused per-residue features, MLP head with ΔΔG = score(mut) − score(wt)).
2. Core motivation: strong in-distribution performance on Megascale does not translate to out-of-distribution (OOD) proteins; datasets are heavily label-imbalanced (stabilizing mutations are rare, ~4–13% across common benchmarks), and predictors show persistent forward–reverse bias on paired-mutation tests (Ssym).
3. The framework combines three losses that target different failure modes: (i) Balanced MSE (BMC) to counter ΔΔG label imbalance, (ii) a Siamese anti-symmetric regularizer to encourage thermodynamic reversibility, and (iii) a new OOD-margin consistency loss that penalizes prediction sensitivity to small perturbations of the per-position fused representation.
4. Headline OOD results across 3 seeds and 11 benchmarks: Spearman on S669 improves from 0.486 to 0.540 (σ=0.002), and on S461 from 0.653 to 0.711. Additional smaller gains are reported on S8754, S2648, S4346, and Ssym-direct; performance drops modestly on in-distribution Megascale-test (0.749 → 0.713), interpreted as a robustness tradeoff.
5. BMC is used as a distribution-aware regression objective with a learnable noise scale, designed to increase gradient pressure on underrepresented ΔΔG regions (especially stabilizing tail) rather than letting MSE/Huber be dominated by neutral/destabilizing examples.
6. The Siamese anti-symmetric loss is applied by evaluating both wt→mut and mut→wt with shared weights and penalizing (f→ f←)^2. Ablations suggest it contributes additively with BMC on the hardest OOD sets, but it can hurt ΔTm benchmarks (e.g., S571), consistent with ΔTm not obeying the same magnitude constraints as ΔΔG.
7. The OOD-margin loss is a representation-stability regularizer: add small Gaussian noise to the fused residue representation after the encoder forward pass, re-run only the MLP head, and penalize (ŷclean − ŷnoisy)^2. It adds ~10% per-step training cost and shows a localized optimum around noise scale σ≈0.20 (too large degrades both OOD gains and in-distribution fit).
8. Mechanistic diagnostic on Ssym: anti-symmetric training does not eliminate systematic forward–reverse bias (offsets remain ~0.3–0.4 kcal/mol). The paper argues gains mainly come from implicit regularization/optimization dynamics rather than strict enforcement of thermodynamic constraints; even an explicit bias-corrected anti-symmetry loss reduces bias but does not improve OOD Spearman.
9. Practical engineering angle: for retrieving rare stabilizing mutations (ΔΔG ≤ −0.5) on S669, the combined objective improves top-50% stabilizing recall (0.659 → 0.685), suggesting better candidate yield in typical screening-style prioritization where the stabilizing tail matters more than average error near neutrality.
10. Negative results help delineate what does not help OOD here: auxiliary multitask supervision with K50 adds little (ΔΔG already highly correlated with K50), and ProteinMPNN-based structural relaxation/perturbation features did not improve key wild-type-based OOD sets (S669/S461), reinforcing that optimization behavior itself can be a bottleneck.
💻Code: github.com/shiv-ram-repo/con…
📜Paper: arxiv.org/abs/2606.08100
#ProteinStability #DDG #ProteinEngineering #ComputationalBiology #MachineLearning #FoundationModels #OODGeneralization #RepresentationLearning #ESM2 #ProteinMPNN
5
28
1,689

Jun 10
General protein diffusion is not enough for antibody engineering!
RFDiffusion3 is SOTA for protein generation. But antibodies require precise optimization of CDR loop geometry, antigen-contact interfaces, and structural specificity. Generic diffusion spreads capacity across the whole protein — exactly the wrong approach for CDR design.
We fine-tuned it with CDR-only noising, CDR-only loss computation, 2x interface weighting, and selective parameter tuning.
Training: 5,017 curated antigen-antibody complexes synthetic data, 159.8M trainable parameters, 18 transformer blocks.
Results: 6.5x better CDR RMSD. 3.1x more strict-passing candidates vs base RFD3.
Visit us at boltzmann.co and accelerate your research journey with us!
#AntibodyDesign #ProteinEngineering #DiffusionModel #CDR #AIBiology #Biopharma #DrugDiscovery
7
325

Jun 9
🔗BioPipelines: Accessible Computational Protein and Ligand Design for Chemical Biologists. doi.org/10.34133/csbj.0129
📚CSBJ - A Science Partner Journal: spj.science.org/journal/csbj
@CSB_Journal @SPJournals @aaas #ProteinEngineering #StructuralBiology #Bioinformatics #AlphaFold #AI

ALT 🔗 BioPipelines: Accessible Computational Protein and Ligand Design for Chemical Biologists. Computational and Structural Biotechnology Journal (CSBJ). DOI: https://doi.org/10.34133/csbj.0129 📚 CSBJ - A Science Partner Journal: https://spj.science.org/journal/csbj
5
16
8,404

Custom protein binders are crucial for treating disease, but discovering them usually takes months.
Recently published in PNAS, new research from the Bryan Dickinson lab has introduced an innovative new platform that completely rewrites that timeline.
Spearheaded by researchers Joshua A. Pixley and Matthew Styles, the PANCS-spec-Binders platform screens billions of synthetic variants to isolate high-fidelity custom binders in just days. By eliminating a notorious biochemical bottleneck and removing the need for secondary screenings, their work opens the door for rapid, highly targeted therapeutic discovery.
Read the full story behind this significant breakthrough and the culture of innovation at UChicago driving it home: shorturl.at/ss9Ba
#Biotech #ProteinEngineering #DrugDiscovery #UChicago #Chemistry #PNAS #ChemicalBiology

2
242
Jun 9
🔗 Optimized Quantitative Bacterial Two-Hybrid (qB2H) for Protein–Protein Interaction Assessment. doi.org/10.34133/csbj.0098
📚 CSBJ - A Science Partner Journal: spj.science.org/journal/csbj
@CSB_Journal @SPJournals @aaas
#ProteinEngineering #SyntheticBiology #StructuralBiology
ALT 🔗 Optimized Quantitative Bacterial Two-Hybrid (qB2H) for Protein–Protein Interaction Assessment. Computational and Structural Biotechnology Journal (CSBJ). DOI: https://doi.org/10.34133/csbj.0098 📚 CSBJ - A Science Partner Journal: https://spj.science.org/journal/csbj
5
3
141

We are looking for a postdoctoral candidate with experience in display techniques to co-write a fellowship application around a research program currently being developed within the team.
#postdoc #phagedisplay #antibodyengineering #proteinengineering #MSCA #ARCFellowship #CNRS

1
110

🚨 Scientists just figured out how to reverse aging with AI.
(Video & Sources below 👇)
By combining Yamanaka factors with AI-designed protein engineering, researchers made cell reprogramming far more effective — pointing to a future where damaged or aged human cells could be restored to a much younger biological state.
This is huge for longevity and regenerative medicine. 🧬🔬
The breakthrough comes from a collaboration between OpenAI and Retro Biosciences (a longevity startup backed by Sam Altman).
They created GPT-4b micro — a specialized miniature AI model trained on protein sequences, biological data, and 3D structures. It redesigned key Yamanaka factors (like SOX2 and KLF4) into enhanced variants (e.g., RetroSOX and RetroKLF).
Results?
50x higher expression of stem cell reprogramming markers vs. wild-type controls.
Dramatically improved DNA damage repair.
• Much higher efficiency (original process was <0.1% success over weeks; new versions hit 30% in tests).
Full details in the official announcement:
openai.com/index/acceleratin…
More coverage:
finance.yahoo.com/news/ai-ju…
Background:
Yamanaka factors (Oct4, Sox2, Klf4, c-Myc) earned Shinya Yamanaka a Nobel Prize for turning adult cells into induced pluripotent stem cells (iPSCs), effectively resetting cellular age. But efficiency was a big bottleneck — until AI stepped in.
This accelerates everything from treating age-related diseases to potential partial reprogramming therapies.
Early days, but incredibly promising.
Watch more:
• Explainer video on the breakthrough: youtube.com/watch?v=ztlSid-O…
• Interview on the OpenAI/Retro partnership: youtube.com/watch?v=dwWjpKzB…
What do you think — is AI about to crack biological aging? Drop your thoughts below! 👇
#YamanakaFactors #ReverseAging #Longevity #AIinBiotech #StemCellResearch #ProteinEngineering #RetroBiosciences #OpenAI
15
39
2,577
🧬💻 Exploring protein mutations just got easier with new computational tools!
📖Read more at: mdpi.com/2674-0583/4/2/11
#ProteinEngineering #ComputationalBiology #StructuralBiology #Bioinformatics #ProteinModeling #Biotechnology #MolecularBiology #DrugDiscovery #ProteinScience
6

Call for AI/ML-Guided Microbial & Protein Engineering Proposals Now Open!
@ICGEBNewDelhi #Biofoundry, invites Expressions of Interest from academia, startups, and industry for AI/ML-driven engineering of E. coli and yeast strains, and protein/enzyme design projects of industrial relevance. Applications are accepted on a rolling basis and reviewed quarterly. Scan the code for more details !
#ICGEB #DBTIndia #Biofoundry #RollingCall #AIinBiotech #SyntheticBiology #Biomanufacturing #ProteinEngineering #StrainEngineering #Yeast #Ecoli #DBTL
2
3
190
Heuristic multi-site optimization for protein sequence design using Masked Protein Language Models @PLOSCompBiol
1 ProtHMSO is a heuristic protein sequence design framework that uses masked protein language models (mainly ESM-2) to propose context-aware, multi-site substitutions, aiming to escape local optima and reduce the “invalid/destabilizing” variants common in blind random mutagenesis.
2 Key idea: mask one or multiple target positions, let the ProtLM output substitution probabilities conditioned on the entire sequence context, and use top-k (k=3 worked best) candidate substitutions to generate a small, high-potential mutant set for fitness scoring—shrinking combinatorial search while keeping evolutionary/biophysical plausibility.
3 The multi-site masking is central: substitutions for all masked sites are predicted synchronously from global context, so probabilities update as the sequence changes. This provides a zero-shot way to capture epistasis (synergistic residue interactions) without explicit structural supervision or task-specific training.
4 ProtHMSO is positioned as both (a) a standalone iterative optimizer and (b) a plug-in mutation operator that can replace random exploration steps inside classic search methods, improving convergence and sample-efficiency.
5 GA-HMSO: integrates ProtHMSO into a genetic algorithm by replacing random mutation with ESM-2-guided mutation, and uses a multi-objective fitness (sum of predictor scores). A dynamic schedule (higher mutation early, lower later) improved exploration–exploitation balance and avoided premature convergence.
6 MCTS-HMSO: integrates ProtHMSO into Monte Carlo Tree Search by using ESM-2 probabilities to guide expansion. It also introduces grouping of child nodes by mutation position (choose site first, then substitution), mitigating the wide-and-shallow tree problem in high-dimensional sequence action spaces.
7 AMP benchmark (DBAASP-derived; three challenging cases): across 1–5 site mutations, ProtHMSO consistently improved antimicrobial metrics (PAMP, PMIC) over random mutagenesis, while also improving structural plausibility proxies (higher ESMFold pLDDT, lower ProGen2 perplexity). Notably, random multi-site mutation degraded plausibility as sites increased, while ProtHMSO improved it.
8 ProteinGym benchmark (long proteins; single-site focus for scalability): on the Clinical substitution benchmark, ProtHMSO produced about 2x more non-pathogenic variants than random mutation at matched library sizes (10/50/100 variants per sequence). On DMS benchmarks (including targeted functional-site mutagenesis), ProtHMSO showed higher enrichment of experimentally top-ranked high-fitness mutants (top-10/20/50 overlaps) for both single- and two-site settings.
9 Practical framing: ProtHMSO acts as a high-throughput “candidate narrowing” layer—reducing millions of possibilities to tens/hundreds—while remaining compatible with adding stricter downstream filters (e.g., Rosetta/MD) in a coarse-to-fine pipeline.
💻Code: github.com/chen-bioinfo/Prot…
📜Paper: doi.org/10.1371/journal.pcbi…
#ProteinDesign #ProteinEngineering #ProteinLanguageModels #ESM2 #DirectedEvolution #GeneticAlgorithms #MCTS #AntimicrobialPeptides #ProteinGym #ComputationalBiology

1
25
2,058
AlloGen: Conformation-Selective Binder Generation with Differential State Scoring
1. AlloGen targets a core limitation in protein binder design: optimizing affinity to a single receptor structure can yield binders that engage both active/inactive (apo/holo) states, providing little functional specificity for allosteric systems (kinases, nuclear receptors, GPCRs).
2. The framework decouples generation from evaluation: any backbone generator proposes candidates for the desired state, then a learned scorer Qθ ranks or guides designs by a differential selectivity margin between goal (holo) and undesired (apo) conformations.
3. Qθ is an SE(3)-invariant interface graph transformer that scores receptor–binder interface geometry in a rigid-motion-invariant way, using interface graphs (8 Å cutoff) with residue-local frames, geometric edge features (distance RBFs, directions, relative orientations), and optional ESM-2 embeddings.
4. Training uses a two-phase curriculum to avoid degenerate “ignore-the-conformation” solutions: Phase 1 regresses to DockQ (interface quality grounding), then Phase 2 applies paired InfoNCE fine-tuning on (holo, apo, binder) triplets with cross-target negatives to force true conformational discrimination rather than receptor identity bias.
5. On 8 held-out OOD targets, Qθ shows consistent rank correlation with DockQ (mean Spearman ρ ≈ 0.520), while contact/energy proxies (PRODIGY, interface size, edge density) largely fail to track docking quality and cannot provide a differential state signal.
6. Qθ appears to encode target- and conformation-specific information: cross-target scoring shows strong diagonal dominance (designs score best on their intended target/state), and on calmodulin it produces a monotonic score increase along an interpolated apo→holo conformational path, suggesting it learns a continuous landscape rather than a binary label.
7. Because Qθ is differentiable and generator-agnostic, it supports multiple integration modes without retraining generators: passive best-of-K reranking and active guidance (classifier guidance, twisted diffusion sampling, SMC resampling, and post-generation Langevin refinement).
8. Across 15 generator×guidance combinations (RFdiffusion, PXDesign, Proteina-ComplexA), resampling-based guidance (TDS/SMC) is broadly strongest; Langevin refinement helps structure-only generators but can harm sequence-aware priors (e.g., PXDesign), emphasizing that guidance interacts with generator assumptions.
9. Experimental validation on calmodulin (a challenging ~30 Å apo↔holo rearrangement) supports the computational selectivity signal: 5/10 synthesized de novo peptides bound holo CaM (KD 46.6 nM to 1.06 µM) with no detectable apo binding, while a low-∆q negative control showed no binding—linking predicted differential scoring to measurable state specificity.
10. The study positions conformational selectivity as a learnable, transferable design objective: a modular scorer trained on paired states can retrofit existing binder-generation pipelines to design molecules that recognize functional states rather than static structures.
💻Code: huggingface.co/ChatterjeeLab…
📜Paper: arxiv.org/abs/2606.05474
#ComputationalBiology #ProteinDesign #GenerativeAI #MachineLearning #StructuralBiology #Allostery #ProteinEngineering #DiffusionModels #GNN #Calmodulin

7
35
2,392

Jun 3
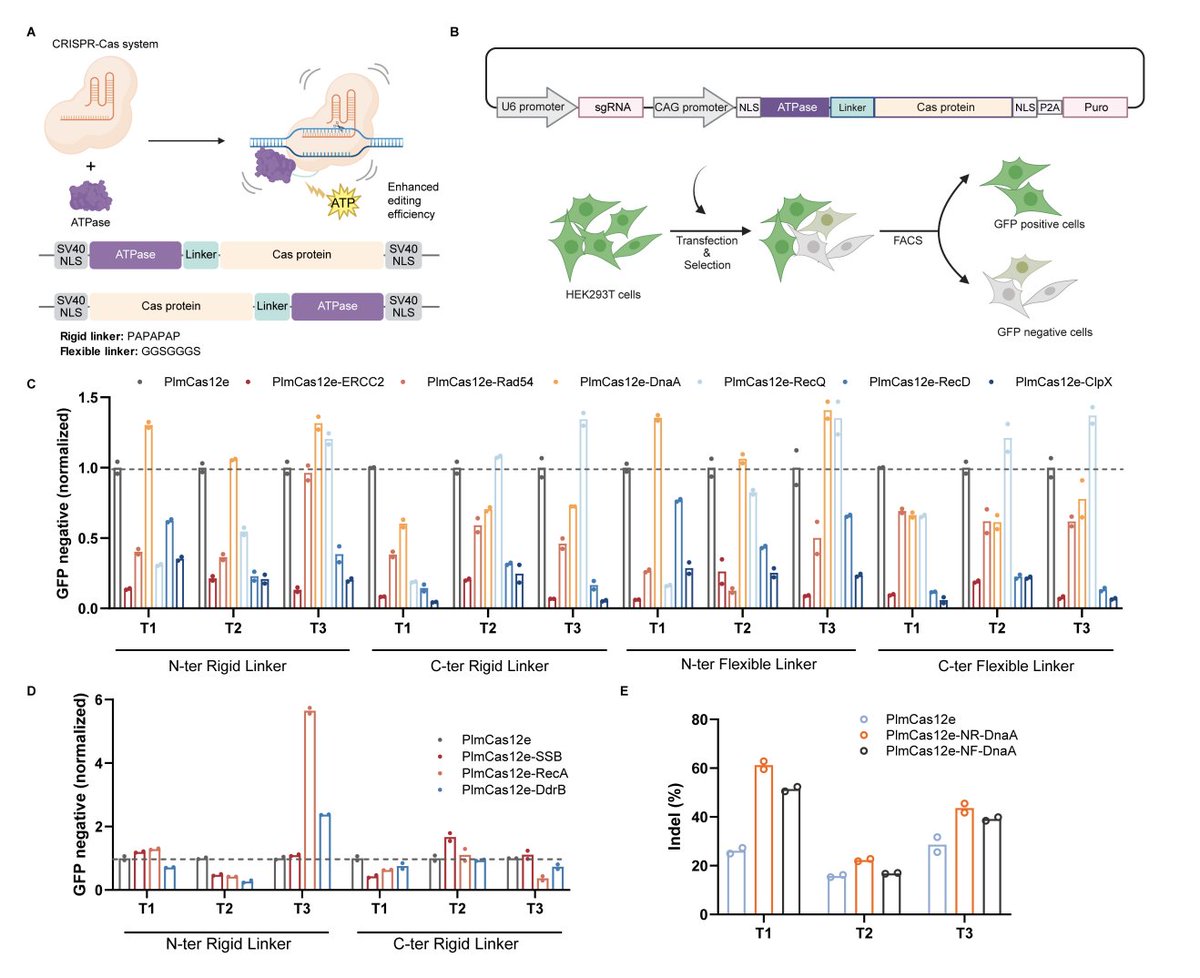
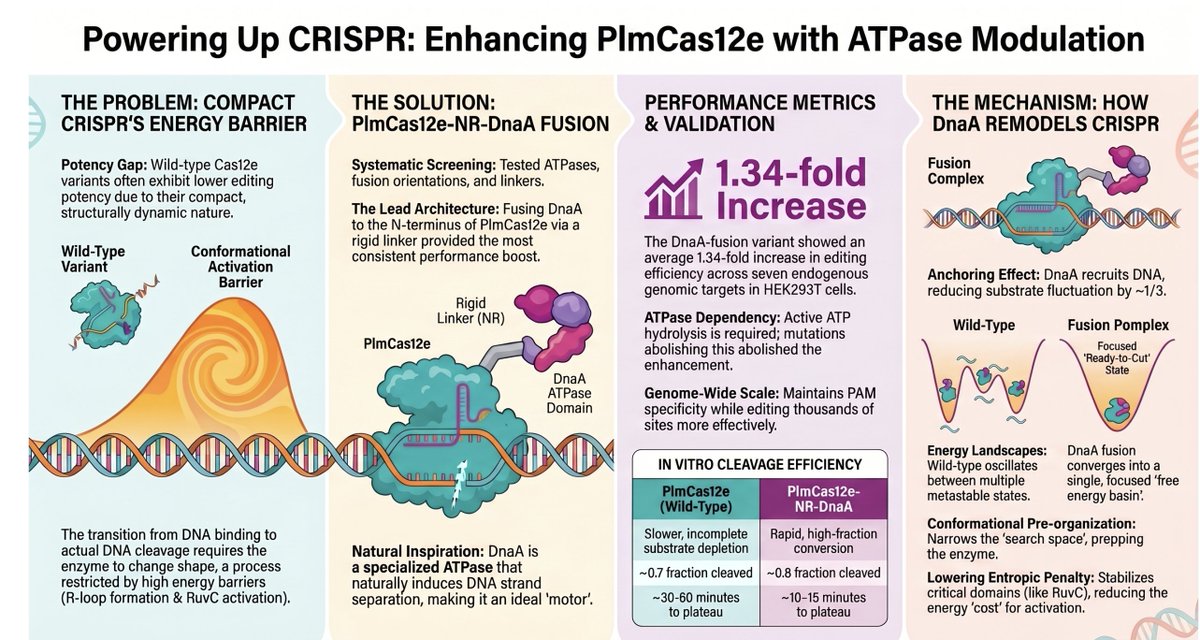
🧬 Compact CRISPR nucleases are small — but often slow to activate. A new strategy just fixed that.
New preprint from Liu Junjie lab (Tsinghua): fusing ATPase DnaA to PlmCas12e (CasX) via a rigid N-terminal linker overcomes the conformational activation barrier.
⚡ 1.34× average editing boost across 7 endogenous loci in HEK293T
📈 More effective genome targets detected by GenomePAM/GUIDE-seq
🔬 MD simulation: single free-energy basin vs. rugged landscape in WT
❌ ATP hydrolysis mutant: activity drops below wild-type — mechanism confirmed
Liu lab, Tsinghua | Preprint
doi.org/10.65215/LTSpreprint…
#CRISPR #Cas12e #CasX #ProteinEngineering #GeneEditing

5
15
870