
Jun 11
AI Is Rewriting mRNA Drug Design
The success of COVID-19 mRNA vaccines proved that messenger RNA can become a transformative therapeutic platform. Yet one of the field’s biggest challenges remains unresolved:
How do we design the optimal mRNA sequence among billions of possibilities?
A new review in Journal of Advanced Research highlights how artificial intelligence is rapidly becoming the engine that drives next-generation mRNA therapeutics.
Unlike conventional drugs, mRNA performance depends heavily on sequence architecture:
• 5′ untranslated region (5′UTR)
• Coding sequence (CDS)
• 3′ untranslated region (3′UTR)
• Secondary structure
• Codon usage patterns
Even when two mRNAs encode exactly the same protein, differences in sequence design can dramatically alter:
✓ Translation efficiency
✓ Stability
✓ Immunogenicity
✓ Tissue-specific expression
The review describes a major paradigm shift.
Generation 1: Rule-based optimization
Historically, mRNA engineering relied on:
• Codon adaptation indices
• Kozak sequence tuning
• Empirical UTR selection
• Trial-and-error screening
These approaches explore only tiny regions of an enormous design space.
For example, the synonymous coding space of the SARS-CoV-2 spike protein exceeds 10⁶³² possible mRNA sequences.
Generation 2: AI prediction models
Deep-learning systems such as:
• Optimus 5-Prime
• UTR-LM
• CodonBERT
• mRNABERT
learn sequence–function relationships directly from large experimental datasets.
Rather than relying on hand-crafted rules, these models predict:
• Ribosome loading
• Translation efficiency
• mRNA half-life
• Protein expression output
Generation 3: AI-generated mRNA
The most exciting development is the rise of generative design.
Instead of evaluating existing sequences, AI can now create entirely new ones.
Examples include:
🧬 UTRGAN
🧬 Smart5UTR
🧬 PARADE
🧬 GEMORNA
These systems generate synthetic UTRs and coding sequences optimized for specific objectives such as:
• High expression
• Increased stability
• Reduced immunogenicity
• Cell-type specificity
Some AI-designed UTRs produced:
🚀 Up to 34-fold increases in translation efficiency
🚀 Nearly 100-fold higher vaccine-induced antibody responses
compared with conventional designs.
The next frontier: coordinated design
The review argues that the field is moving beyond isolated optimization of individual sequence elements.
Current efforts increasingly focus on:
5′UTR CDS 3′UTR co-design
as a unified system.
Models such as:
• LinearDesign2
• GEMORNA
• mRNABERT
attempt to optimize the entire transcript simultaneously rather than treating each region independently.
This matters because translation, stability, structure, and immunogenicity emerge from interactions across the full-length mRNA molecule.
Why this matters
The future of mRNA medicine may resemble modern protein design.
Instead of manually optimizing sequence elements, researchers will specify desired properties:
✓ High expression
✓ Long half-life
✓ Low innate immune activation
✓ Liver targeting
✓ Efficient LNP delivery
and AI systems will generate candidate mRNAs automatically.
The authors envision a future built around:
• Foundation models for RNA biology
• Multi-objective optimization
• Generative AI
• Closed-loop design-build-test-learn platforms
where computational models and experimental validation continuously improve each other.
If protein engineering was transformed by AlphaFold and generative biology, mRNA therapeutics may be approaching a similar inflection point.
The next blockbuster mRNA drug may be designed not by manual codon tuning—but by AI.
Reference
Shi Y, Zeng C, Sheng X, et al.
Transforming mRNA drug design with AI: From UTR and codon optimization to coordinated design.
Journal of Advanced Research (2026)
DOI: 10.1016/j.jare.2026.06.013
#mRNA #ArtificialIntelligence #GenerativeAI #CodonOptimization #UTRDesign #RNAEngineering #DrugDiscovery #BioAI #PrecisionMedicine #JournalOfAdvancedResearch

1
6
320

4 Dec 2025
Harnessing Hypergraphs in Geometric Deep Learning for 3D RNA Inverse Folding
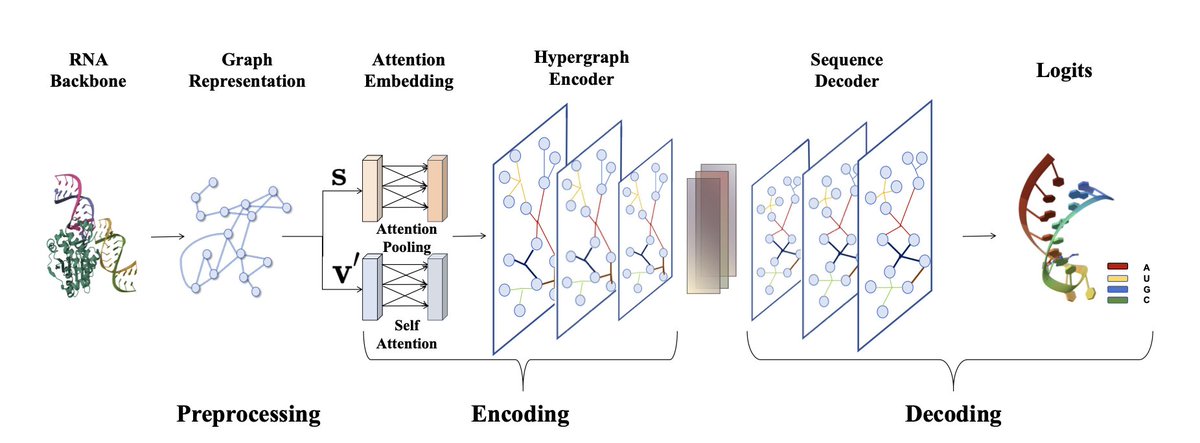
1. This groundbreaking study introduces HyperRNA, a novel framework leveraging hypergraphs to solve the complex RNA inverse folding problem. The model uses an encoder-decoder architecture to generate RNA sequences that can fold into desired secondary structures, addressing a key challenge in RNA design.
2. The core innovation lies in the use of hypergraphs, which capture higher-order dependencies and complex interactions between nucleotides. This is a significant upgrade from traditional graph neural networks, enabling more accurate modeling of RNA's intricate structure and dynamics.
3. HyperRNA's architecture includes a preprocessing stage that constructs graph structures from RNA backbones, an encoding stage that embeds these graphs using attention mechanisms, and a decoding stage that generates RNA sequences autoregressively.
4. Experiments on the PDBBind and RNAsolo datasets demonstrate HyperRNA's superior performance, achieving higher RNA recovery rates and structural accuracy compared to existing methods. The model also shows strong generalization across diverse RNA sequences.
5. Ablation studies highlight the importance of integrating both scalar and vector features in the attention embedding module, which is crucial for capturing essential structural dependencies and improving sequence prediction accuracy.
6. The study underscores the potential of hypergraph-based methods in advancing RNA design, offering a robust framework for more accurate and diverse RNA structure modeling. Future work may explore further optimizations and broader applications in RNA-related fields.
📜Paper: arxiv.org/abs/2512.03592v1
#RNAInverseFolding #Hypergraphs #GeometricDeepLearning #Bioinformatics #RNAEngineering
3
2
21
1,511
2 Dec 2025
Generative Inverse Design of RNA Structure and Function with gRNAde
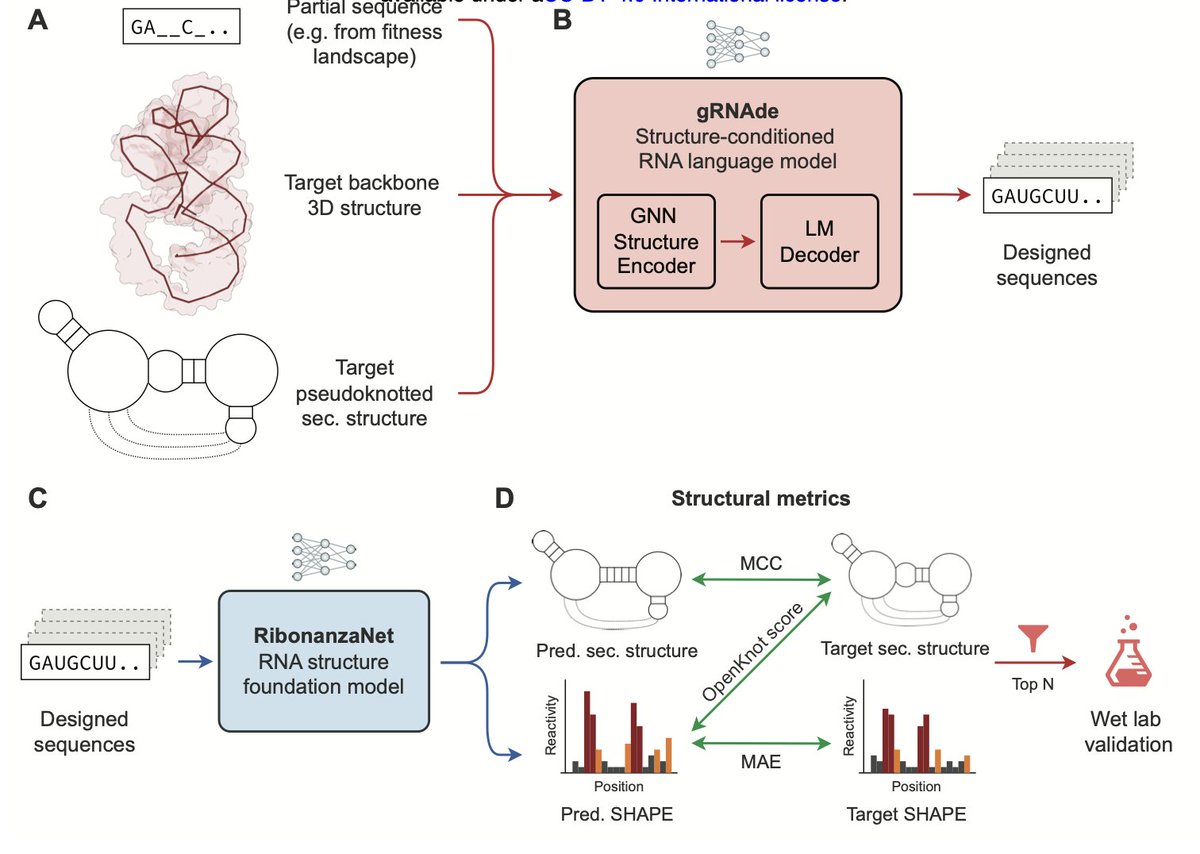
1. A novel study introduces gRNAde, a high-throughput AI pipeline for designing RNA molecules with bespoke 3D structures and functions. This pipeline leverages deep learning to generate RNA sequences that can fold into complex structures, including challenging pseudoknots, matching the success rates of human experts in a community-wide competition.
2. gRNAde integrates a structure-conditioned RNA language model with computational screening to efficiently identify optimal RNA sequences. It demonstrated superior performance in designing functional RNA polymerase ribozymes, discovering highly active variants with up to 20% sequence divergence from the wild type, which were previously inaccessible through rational design or directed evolution.
3. The study highlights gRNAde’s ability to perform large-scale generative “jumps” in sequence space, identifying functional RNA variants far beyond the reach of conventional methods. This capability significantly accelerates the exploration of RNA sequence space and paves the way for fully programmable RNA catalysts.
4. gRNAde’s success in both structural and functional RNA design underscores its potential to revolutionize RNA engineering. The pipeline is open-source, enabling broader community use and further advancements in synthetic biology and biotechnology.
📜Paper: biorxiv.org/content/10.1101/…
#RNAEngineering #DeepLearning #SyntheticBiology #Biotechnology #RNADesign
5
18
5,815
29 Nov 2025
RNA–X: Modeling RNA interactions to design binder RNA and simultaneously target multiple molecules of different types
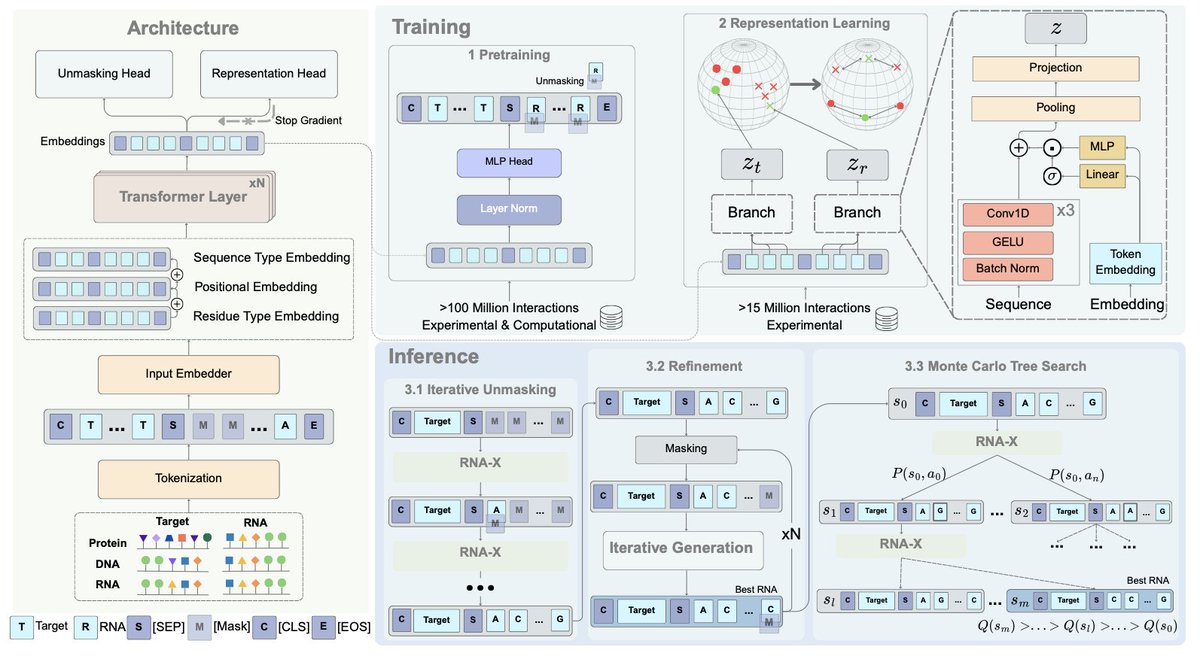
1. A groundbreaking study introduces RNA–X, the first RNA interaction foundation model capable of designing RNA sequences that can target proteins, other RNA molecules, and DNA simultaneously. This innovation significantly expands the potential for RNA-based therapeutic design.
2. RNA–X employs masked language modeling to learn joint representations of RNA and target molecules, enabling the generation of RNA sequences with desired binding properties. The model demonstrates superior performance in protein targeting compared to existing state-of-the-art methods.
3. A key innovation is the ability to design RNA sequences for multiple targets in a single run. This was demonstrated by creating a guide RNA that simultaneously binds to bacterial DNA and the Cas9 protein, achieving binding energy comparable to wild-type sequences.
4. The model's effectiveness is validated through extensive experiments, including molecular dynamics simulations and affinity predictions. RNA–X shows strong binding potential even for targets without prior interaction data, such as the SDAD1 protein.
5. Despite having significantly fewer parameters than existing RNA foundation models, RNA–X outperforms them in downstream tasks like siRNA efficacy and sgRNA on-target knockout predictions. This highlights the efficiency and versatility of the model.
6. The study emphasizes the importance of representation learning, using techniques like whitening and uniformity to enhance the model's ability to capture biological diversity. This results in more informative embeddings and improved performance.
7. RNA–X's architecture includes a shared transformer backbone with sequence-type, positional, and residue-type embeddings. The model is trained on over 100 million RNA–target interactions, ensuring robustness and generalizability.
📜Paper: biorxiv.org/content/10.1101/…
#RNAEngineering #ComputationalBiology #TherapeuticDesign #AIinBiology
6
28
2,168
18 Nov 2025
Geometric Algebra-Enhanced Bayesian Flow Network for RNA Inverse Design
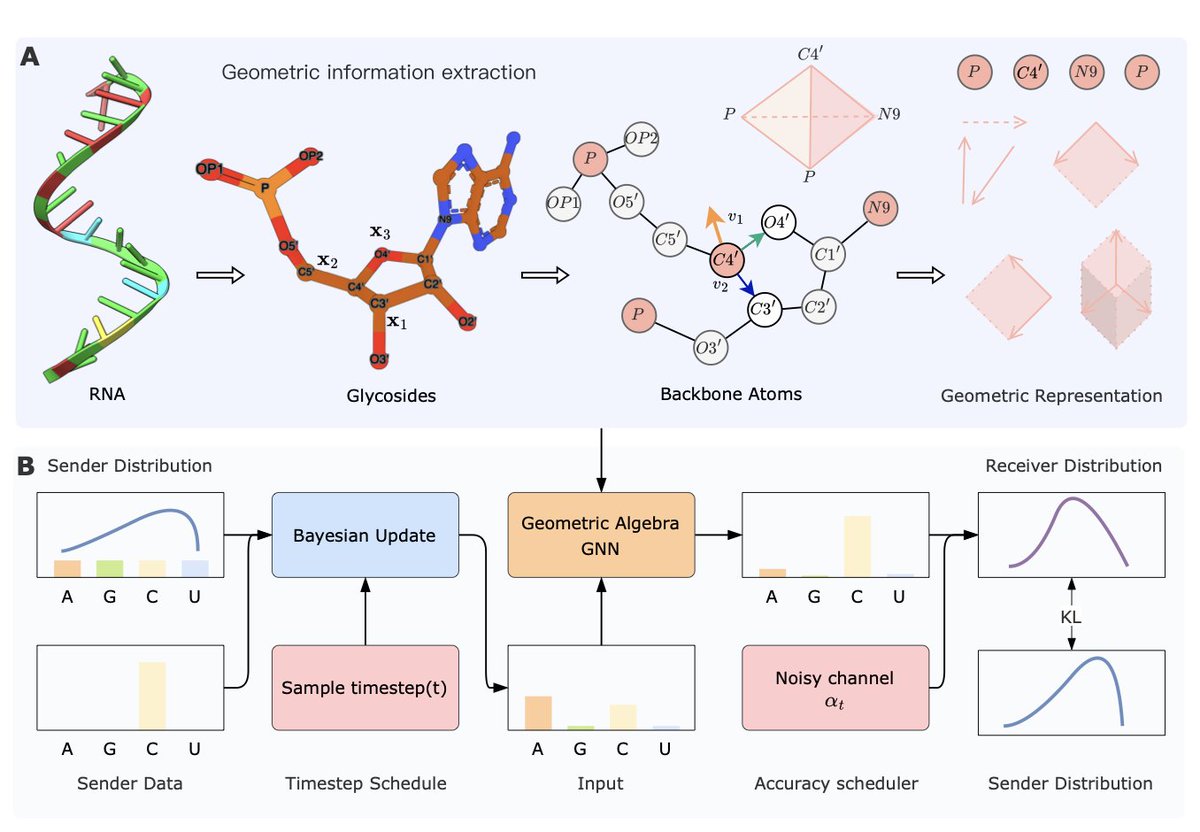
1. The paper introduces RBFN, a novel method for RNA inverse design that combines geometric algebra with Bayesian Flow Networks to generate RNA sequences from 3D structures. This approach addresses the challenge of designing RNA sequences that can fold into specific 3D structures, a critical task in RNA therapeutics and synthetic biology.
2. A key innovation is the use of geometric algebra to enhance the modeling of RNA's 3D structures. By encoding structural information into multivectors, the method captures complex geometric relationships, enabling more accurate and flexible RNA design compared to traditional methods.
3. The Bayesian Flow Network component allows for distribution-based sequence generation, aligning nucleotide distributions rather than generating discrete sequences directly. This probabilistic approach improves the model's ability to explore diverse sequence possibilities and enhances overall design efficiency.
4. RBFN proposes a new time-step sampling distribution tailored for RNA sequences, focusing on the transition from initial to target distributions. This strategy improves the model's global generation ability, particularly important given the limited diversity of RNA nucleotides (A, C, G, U).
5. Extensive experiments demonstrate RBFN's superior performance over state-of-the-art methods, including gRNAde and Rosetta, in both single-state and multi-state RNA design benchmarks. The results highlight significant improvements in sequence recovery rates and structural consistency metrics.
6. The study also includes ablation experiments that validate the contributions of geometric algebra and the new time-step sampling distribution. These components are shown to be crucial for achieving high-quality RNA sequence design with consistent structural properties.
📜Paper: openreview.net/pdf/daf45e7e0…
#RNAInverseDesign #GeometricAlgebra #BayesianFlowNetworks #ComputationalBiology #RNAEngineering
11
50
3,040
6 Oct 2025
RNA Sequence Design and Protein–DNA Specificity Prediction with NA-MPNN
🚀 New preprint from David Baker!🚀
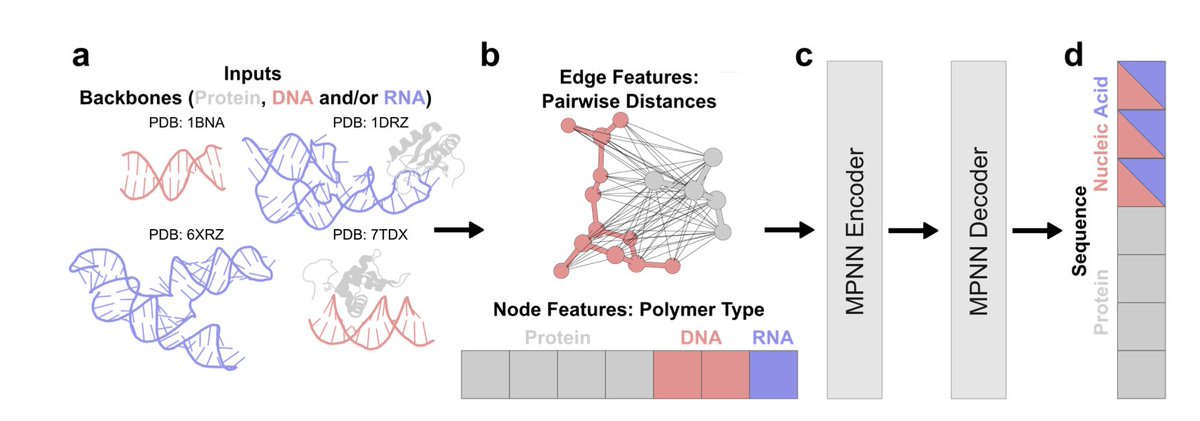
1. A new deep learning model called NA-MPNN has been introduced for RNA sequence design and protein–DNA binding specificity prediction. This model treats proteins, DNA, and RNA within a unified biopolymer graph representation, which is a significant innovation in the field of nucleic acid inverse folding.
2. NA-MPNN outperforms previous methods on RNA sequence design and fixed-dock protein–DNA specificity prediction. It achieves robust sequence recovery across both standalone nucleic acids and protein-bound contexts, with median recoveries of 57.4% for DNA-only, 60.5% for RNA-only, 58.6% for DNA in protein context, and 55.4% for RNA in protein context.
3. The model’s architecture is based on extending ProteinMPNN to enable computation over nucleic acid backbones. It includes nodes that can be either protein residues or DNA or RNA bases, and edges between nodes can be protein–protein, nucleic acid–nucleic acid, or protein–nucleic acid.
4. NA-MPNN uses a unified graph architecture for all polymers but is trained on two task-specific models: a design model for backbone-conditioned sequence design and a specificity model for fixed-dock protein–DNA binding preferences.
5. The specificity model utilizes a set of data augmentations tailored to learning position probability matrix (PPM) targets. It focuses on sequence preferences that are induced by protein contacts rather than artifacts of nucleic acid backbone geometry.
6. In the OpenKnot RNA pseudoknot design challenge, NA-MPNN designs had the highest overall consistency measured by experimental chemical footprinting, demonstrating the successful translation from improved in silico accuracy to wet-lab success.
7. For fixed-dock protein–DNA specificity prediction, NA-MPNN reduces the median mean absolute error and cross-entropy relative to DeepPBS despite operating solely on backbone coordinates and omitting protein side-chain atoms.
8. NA-MPNN is expected to be broadly useful for creating the next generation of designed RNA molecules, transcription factors, and genome engineering tools. The unification of protein, DNA, and RNA inverse folding enables additional applications, including RNA-binding protein specificity prediction and backbone-conditioned sequence design of single-stranded DNA.
📜Paper: biorxiv.org/content/10.1101/…
#NucleicAcidDesign #ProteinDNAInteraction #DeepLearning #Bioinformatics #RNAEngineering #GenomeEditing
9
28
2,159
23 Jul 2025
Efficient Design of RNA Sequences with Desired Properties, Structure, and Motifs Using a Grammar Variational Autoencoder
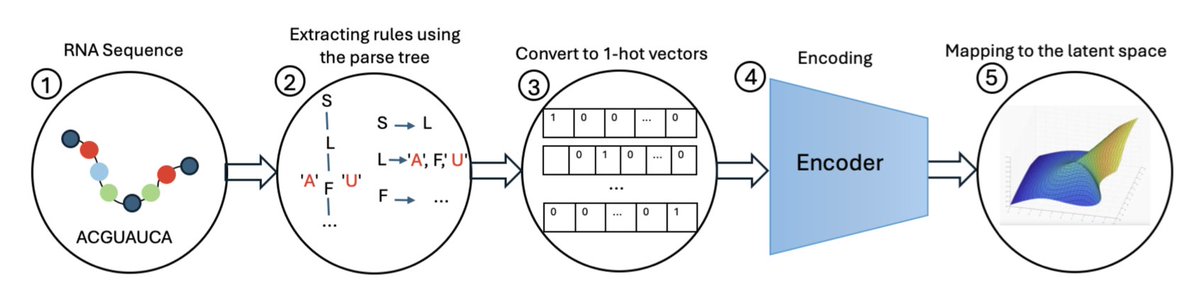
1. This paper introduces the RNA Grammar Variational Autoencoder (RGVAE), a novel deep learning approach for designing RNA sequences with specific target properties, including structural stability, desired motifs, and other constraints. The RGVAE leverages stochastic context-free grammars (SCFGs) to ensure that generated RNA sequences form thermodynamically stable secondary structures.
2. The RGVAE builds on the grammar variational autoencoder (GVAE) framework, incorporating SCFGs to parse RNA sequences into production rules. These rules are then encoded into a continuous latent space, where optimization can be performed efficiently. The optimized latent representations can be decoded back into RNA sequences with desired characteristics.
3. The authors demonstrate that the RGVAE significantly outperforms traditional methods such as randomized design and regular VAEs that do not utilize SCFGs. The model is tested on various practical use cases, including minimizing minimum free energy (MFE), maintaining specific GC content, and incorporating mandatory or forbidden motifs.
4. One of the key innovations of the RGVAE is its ability to optimize RNA sequences in the latent space using Bayesian optimization. This approach allows for efficient exploration of the design space, even for complex constraints involving multiple properties and structural requirements.
5. The study shows that the RGVAE can generate RNA sequences with lower MFE compared to training data and other models, while also satisfying additional constraints such as GC content and specific secondary structures. The flexibility of the model allows it to be applied to a wide range of RNA design scenarios.
6. The authors provide the source code and data used in the study, making it accessible for further research and development in RNA design. The RGVAE represents a significant advancement in the field of bioinformatics, offering a powerful tool for the design of functional RNA molecules with specific properties.
💻Code: github.com/nzarnaghinaghsh/R…
📜Paper: arxiv.org/abs/2507.15912
#RNAEngineering #DeepLearning #Bioinformatics #RNASequences #GrammarVAE
5
799
23 Jul 2025
Efficient Design of RNA Sequences with Desired Properties, Structure, and Motifs Using a Grammar Variational Autoencoder
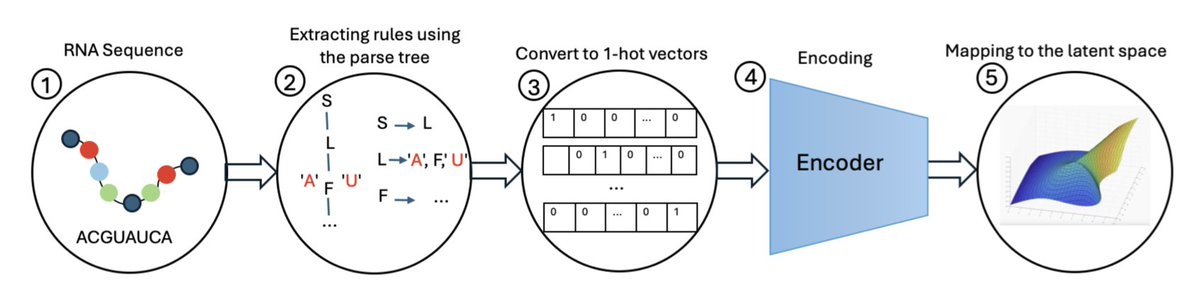
1. This paper introduces the RNA Grammar Variational Autoencoder (RGVAE), a novel deep learning approach for designing RNA sequences with specific target properties, including structural stability, desired motifs, and other constraints. The RGVAE leverages stochastic context-free grammars (SCFGs) to ensure that generated RNA sequences form thermodynamically stable secondary structures.
2. The RGVAE builds on the grammar variational autoencoder (GVAE) framework, incorporating SCFGs to parse RNA sequences into production rules. These rules are then encoded into a continuous latent space, where optimization can be performed efficiently. The optimized latent representations can be decoded back into RNA sequences with desired characteristics.
3. The authors demonstrate that the RGVAE significantly outperforms traditional methods such as randomized design and regular VAEs that do not utilize SCFGs. The model is tested on various practical use cases, including minimizing minimum free energy (MFE), maintaining specific GC content, and incorporating mandatory or forbidden motifs.
4. One of the key innovations of the RGVAE is its ability to optimize RNA sequences in the latent space using Bayesian optimization. This approach allows for efficient exploration of the design space, even for complex constraints involving multiple properties and structural requirements.
5. The study shows that the RGVAE can generate RNA sequences with lower MFE compared to training data and other models, while also satisfying additional constraints such as GC content and specific secondary structures. The flexibility of the model allows it to be applied to a wide range of RNA design scenarios.
6. The authors provide the source code and data used in the study, making it accessible for further research and development in RNA design. The RGVAE represents a significant advancement in the field of bioinformatics, offering a powerful tool for the design of functional RNA molecules with specific properties.
💻Code: github.com/nzarnaghinaghsh/R…
📜Paper: arxiv.org/abs/2507.15912
#RNAEngineering #DeepLearning #Bioinformatics #RNASequences #GrammarVAE
7
775
11 Apr 2025
Capturing Natural Evolution in Function-guided RNA Design via Genomic Foundation Models
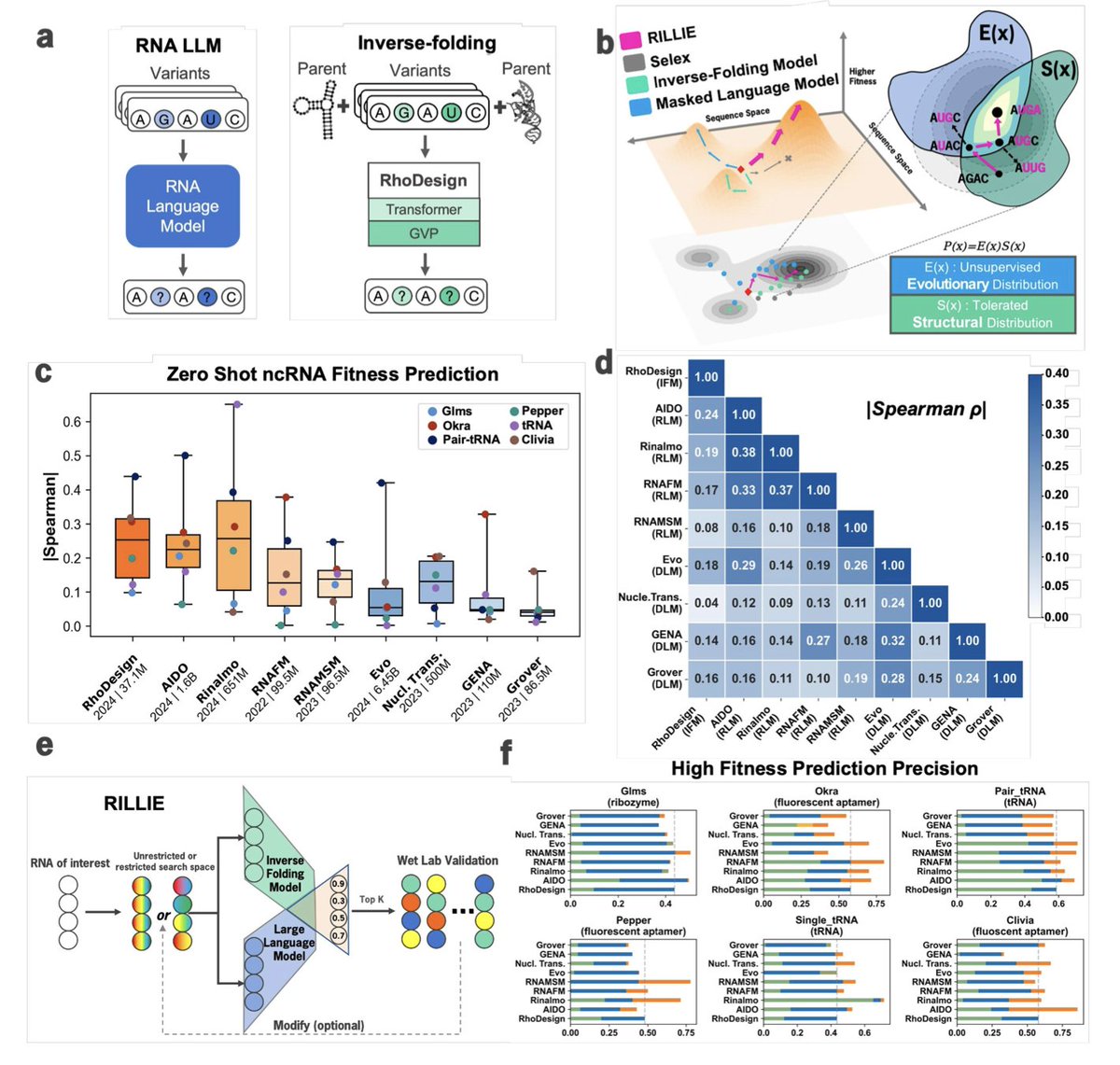
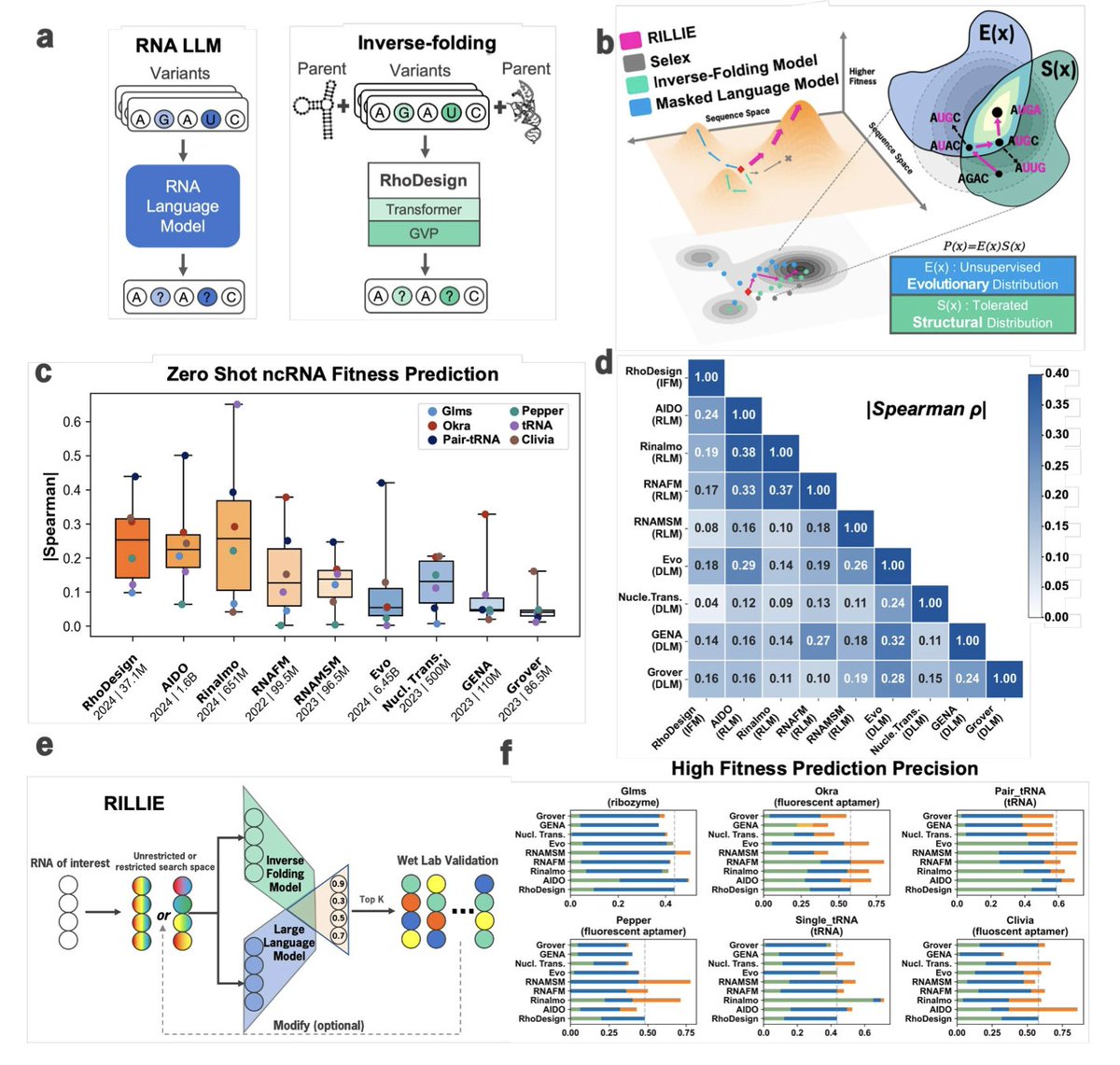
1. This study introduces RILLIE, a zero-shot RNA design framework that combines large language models (LLMs) and inverse folding models (IFMs) to simulate natural evolution and optimize RNA sequences for in vivo function—without any task-specific training.
2. RILLIE integrates AIDO.RNA, a 1.6B-parameter RNA LLM capturing evolutionary plausibility, with RhoDesign, an inverse folding model capturing structural compatibility, forming a “product of experts” model that jointly optimizes for sequence and structure fitness.
3. Unlike traditional SELEX or task-specific ML pipelines, RILLIE operates in a zero-shot setting, rapidly generating RNA variants that maintain natural sequence grammar and structural integrity while enhancing experimental performance.
4. Benchmarking across six diverse DMS datasets (aptamers, tRNAs, ribozymes), RILLIE demonstrates high correlation between model scores and experimental RNA fitness, significantly outperforming both RNA and DNA LLMs alone.
5. Applied to the Broccoli aptamer, RILLIE generated 20 variants in a single round—over half showed increased fluorescence, with B2 achieving a 55% boost in intensity and a 2x improvement in binding affinity, verified via FACS in living HEK cells.
6. For the Pepper aptamer, a two-round directed evolution strategy yielded 40 novel variants, with fluorescence boosts up to 2.6-fold and 3x binding affinity improvement. Over 40% of sequences outperformed wild type, including high-mutation variants with up to 75% sequence divergence.
7. Mutation preference analysis revealed that RILLIE avoids deleterious substitutions (e.g., C5G, U19A) and favors beneficial mutations in variable regions, showing strong alignment with natural selection patterns and high-fitness precision.
8. Importantly, sequences designed with RILLIE retained performance in vivo, demonstrating improved folding and function in HEK cells—a major challenge for aptamers designed solely via in vitro methods like SELEX.
9. RILLIE’s framework can generalize to other RNA classes beyond aptamers. The model was shown to perform well in predicting mutational effects across ribozymes and tRNAs, opening pathways for universal RNA engineering.
10. This work provides the first large-scale evidence that integrating structural and sequence models allows for scalable, evolution-guided, task-agnostic RNA design—enabling a paradigm shift in synthetic biology and RNA therapeutics.
💻Code: github.com/GENTEL-lab/RILLIE
📜Paper: biorxiv.org/content/10.1101/…
#RNAEngineering #SyntheticBiology #RNAaptamers #ZeroShotLearning #LanguageModels #InverseFolding #Bioinformatics #AptamerDesign #RNAtherapeutics #LLM #RILLIE #DirectedEvolution
3
22
1,951
11 Apr 2025
Capturing Natural Evolution in Function-guided RNA Design via Genomic Foundation Models
1. This study introduces RILLIE, a zero-shot RNA design framework that combines large language models (LLMs) and inverse folding models (IFMs) to simulate natural evolution and optimize RNA sequences for in vivo function—without any task-specific training.
2. RILLIE integrates AIDO.RNA, a 1.6B-parameter RNA LLM capturing evolutionary plausibility, with RhoDesign, an inverse folding model capturing structural compatibility, forming a “product of experts” model that jointly optimizes for sequence and structure fitness.
3. Unlike traditional SELEX or task-specific ML pipelines, RILLIE operates in a zero-shot setting, rapidly generating RNA variants that maintain natural sequence grammar and structural integrity while enhancing experimental performance.
4. Benchmarking across six diverse DMS datasets (aptamers, tRNAs, ribozymes), RILLIE demonstrates high correlation between model scores and experimental RNA fitness, significantly outperforming both RNA and DNA LLMs alone.
5. Applied to the Broccoli aptamer, RILLIE generated 20 variants in a single round—over half showed increased fluorescence, with B2 achieving a 55% boost in intensity and a 2x improvement in binding affinity, verified via FACS in living HEK cells.
6. For the Pepper aptamer, a two-round directed evolution strategy yielded 40 novel variants, with fluorescence boosts up to 2.6-fold and 3x binding affinity improvement. Over 40% of sequences outperformed wild type, including high-mutation variants with up to 75% sequence divergence.
7. Mutation preference analysis revealed that RILLIE avoids deleterious substitutions (e.g., C5G, U19A) and favors beneficial mutations in variable regions, showing strong alignment with natural selection patterns and high-fitness precision.
8. Importantly, sequences designed with RILLIE retained performance in vivo, demonstrating improved folding and function in HEK cells—a major challenge for aptamers designed solely via in vitro methods like SELEX.
9. RILLIE’s framework can generalize to other RNA classes beyond aptamers. The model was shown to perform well in predicting mutational effects across ribozymes and tRNAs, opening pathways for universal RNA engineering.
10. This work provides the first large-scale evidence that integrating structural and sequence models allows for scalable, evolution-guided, task-agnostic RNA design—enabling a paradigm shift in synthetic biology and RNA therapeutics.
💻Code: github.com/GENTEL-lab/RILLIE
📜Paper: biorxiv.org/content/10.1101/…
#RNAEngineering #SyntheticBiology #RNAaptamers #ZeroShotLearning #LanguageModels #InverseFolding #Bioinformatics #AptamerDesign #RNAtherapeutics #LLM #RILLIE #DirectedEvolution
4
15
1,748
4 Apr 2025
Integrating experimental feedback improves generative models for biological sequences
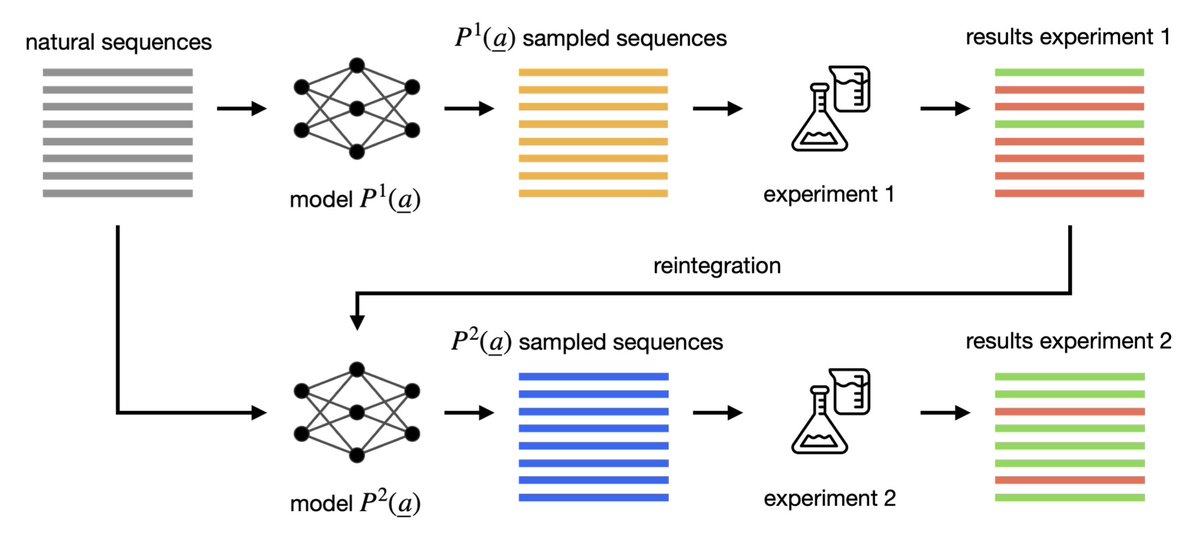
1. This work introduces a likelihood-based reintegration method that incorporates experimental feedback into generative models for RNA and protein sequences, drastically reducing false positives without altering model architecture.
2. Applied to DCA models, the reintegration method penalizes non-functional sequences and rewards functional ones using weighted likelihoods, refining the generative boundaries of viable sequence space.
3. Across multiple RNA families from Rfam, reintegration improved the fraction of predicted functional sequences from ~50% to over 99%, as measured by thermodynamic stability using RNAeval folding free energy.
4. In chorismate mutase (CM) protein design, the percentage of predicted functional sequences increased from 39% to 68% after reintegration, with minimal loss in diversity, validating the approach on real protein fitness landscapes.
5. Experimental validation on Group I intron ribozymes confirmed that reintegrated models generate far more active sequences at higher mutation distances—up to 63.7% activity at 45 mutations, compared to 6.7% for the non-reintegrated model.
6. A variant of the reintegration scheme, REINT BS0, grouped sequences by mutation bins and maintained balance across distance levels, successfully mitigating overfitting to wildtype and extending functional generation up to 65 mutations.
7. Reintegration consistently improves the model’s ability to predict actual fitness (proxy or experimental), showing stronger correlation between model energy and experimental activity post-training.
8. The approach is general and compatible with any generative model (e.g. VAEs, RBMs, PLMs), and it only requires adjusting the data likelihood to reflect experimental results—offering broad utility in data-driven biological design.
9. The key insight is that limitations of generative models are often due to underinformative training data, not model expressivity; refining these models with experimental labels substantially enhances their predictive power.
10. This work offers a practical, principled path to more reliable biomolecular design—by closing the loop between generative prediction and experimental feedback, and doing so in a mathematically tractable way.
📜Paper: arxiv.org/abs/2504.01593
#ProteinDesign #RNAEngineering #GenerativeModels #MachineLearning #SyntheticBiology #Bioinformatics #DCA #MolecularDesign #ComputationalBiology #HighThroughput #RNA #ProteinEngineering
2
11
1,324
4 Apr 2025
Integrating experimental feedback improves generative models for biological sequences
1. This work introduces a likelihood-based reintegration method that incorporates experimental feedback into generative models for RNA and protein sequences, drastically reducing false positives without altering model architecture.
2. Applied to DCA models, the reintegration method penalizes non-functional sequences and rewards functional ones using weighted likelihoods, refining the generative boundaries of viable sequence space.
3. Across multiple RNA families from Rfam, reintegration improved the fraction of predicted functional sequences from ~50% to over 99%, as measured by thermodynamic stability using RNAeval folding free energy.
4. In chorismate mutase (CM) protein design, the percentage of predicted functional sequences increased from 39% to 68% after reintegration, with minimal loss in diversity, validating the approach on real protein fitness landscapes.
5. Experimental validation on Group I intron ribozymes confirmed that reintegrated models generate far more active sequences at higher mutation distances—up to 63.7% activity at 45 mutations, compared to 6.7% for the non-reintegrated model.
6. A variant of the reintegration scheme, REINT BS0, grouped sequences by mutation bins and maintained balance across distance levels, successfully mitigating overfitting to wildtype and extending functional generation up to 65 mutations.
7. Reintegration consistently improves the model’s ability to predict actual fitness (proxy or experimental), showing stronger correlation between model energy and experimental activity post-training.
8. The approach is general and compatible with any generative model (e.g. VAEs, RBMs, PLMs), and it only requires adjusting the data likelihood to reflect experimental results—offering broad utility in data-driven biological design.
9. The key insight is that limitations of generative models are often due to underinformative training data, not model expressivity; refining these models with experimental labels substantially enhances their predictive power.
10. This work offers a practical, principled path to more reliable biomolecular design—by closing the loop between generative prediction and experimental feedback, and doing so in a mathematically tractable way.
📜Paper: arxiv.org/abs/2504.01593
#ProteinDesign #RNAEngineering #GenerativeModels #MachineLearning #SyntheticBiology #Bioinformatics #DCA #MolecularDesign #ComputationalBiology #HighThroughput #RNA #ProteinEngineering

2
9
814
24 Mar 2025
RiboFlow: Conditional De Novo RNA Sequence-Structure Co-Design via Synergistic Flow Matching
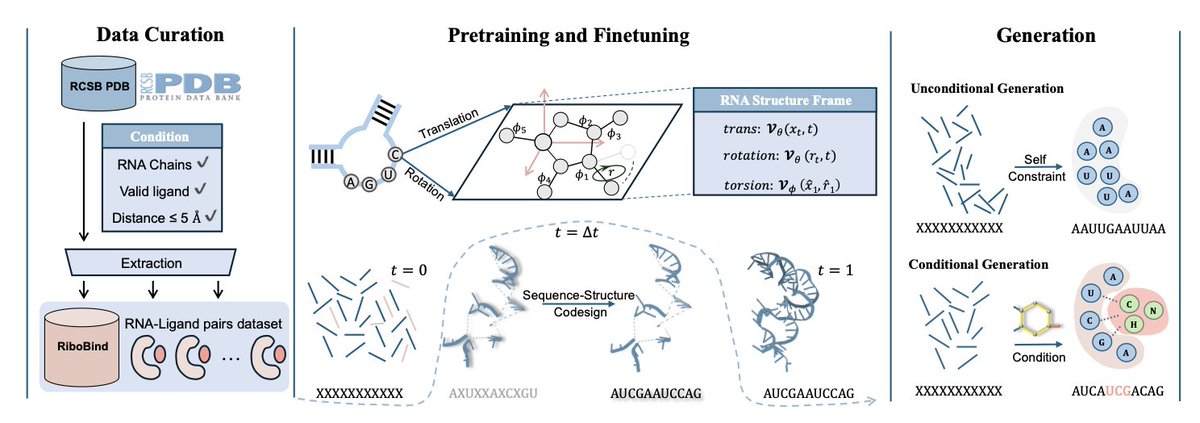
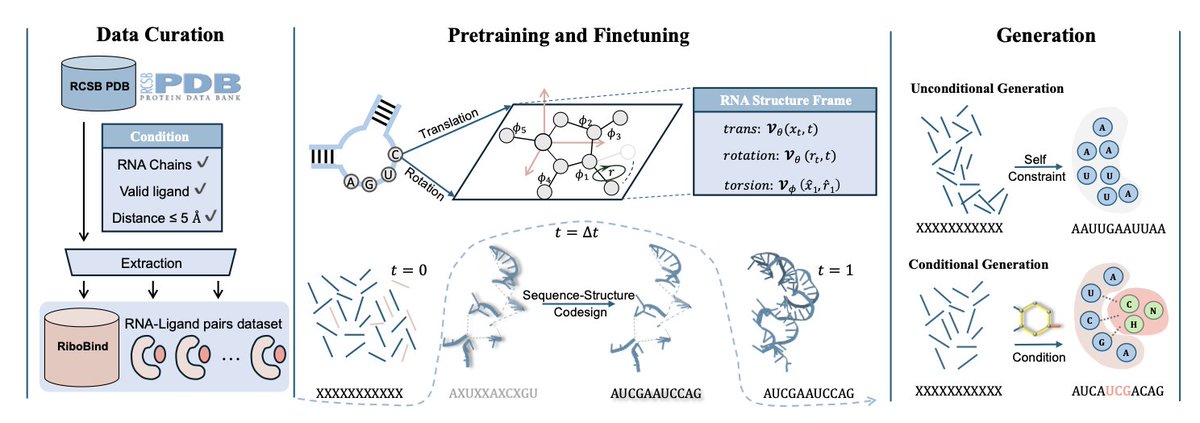
1. This paper introduces RiboFlow, a novel framework designed for de novo RNA sequence-structure co-design. Unlike previous models, RiboFlow incorporates both RNA backbone frames and torsion angles, effectively capturing the dynamic conformations of RNA while ensuring sequence-structure consistency.
2. The model leverages a synergistic flow matching approach to generate RNA structures and sequences based on target molecules. By conditioning on ligand geometry, RiboFlow models RNA-ligand interactions and optimizes binding affinity, outperforming state-of-the-art RNA design methods.
3. A new benchmark dataset, RiboBind, is presented, which includes 1,591 RNA-ligand complexes and 3,012 RNA-ligand pairs. This dataset offers comprehensive structural diversity, providing a valuable resource for training and evaluating RNA-ligand interaction models.
4. RiboFlow achieves a 2.2-fold improvement in the AF3 binding metric and a 50% increase in validity compared to existing models. Its co-design strategy allows the generation of RNA sequences that are structurally valid and have high binding affinity to specific ligands.
5. The model is also capable of controlling the generation process to produce RNAs with desired properties, making it suitable for applications in RNA-based therapeutics, biosensing, and synthetic biology.
6. RiboFlow’s performance is further enhanced through a co-design pre-training strategy, which improves geometric awareness by distilling structural priors from RNA crystal structures.
7. Experimental results demonstrate that RiboFlow surpasses baseline models in terms of structural validity, diversity, novelty, and binding affinity, offering a promising tool for ligand-conditioned RNA design.
📜Paper: arxiv.org/abs/2503.17007
#RiboFlow #RNA #RNAEngineering #Bioinformatics #GenerativeModels #DeepLearning #RNASequenceDesign #MoleculeDesign #MachineLearning #AI
3
12
1,241
24 Mar 2025
RiboFlow: Conditional De Novo RNA Sequence-Structure Co-Design via Synergistic Flow Matching
1. This paper introduces RiboFlow, a novel framework designed for de novo RNA sequence-structure co-design. Unlike previous models, RiboFlow incorporates both RNA backbone frames and torsion angles, effectively capturing the dynamic conformations of RNA while ensuring sequence-structure consistency.
2. The model leverages a synergistic flow matching approach to generate RNA structures and sequences based on target molecules. By conditioning on ligand geometry, RiboFlow models RNA-ligand interactions and optimizes binding affinity, outperforming state-of-the-art RNA design methods.
3. A new benchmark dataset, RiboBind, is presented, which includes 1,591 RNA-ligand complexes and 3,012 RNA-ligand pairs. This dataset offers comprehensive structural diversity, providing a valuable resource for training and evaluating RNA-ligand interaction models.
4. RiboFlow achieves a 2.2-fold improvement in the AF3 binding metric and a 50% increase in validity compared to existing models. Its co-design strategy allows the generation of RNA sequences that are structurally valid and have high binding affinity to specific ligands.
5. The model is also capable of controlling the generation process to produce RNAs with desired properties, making it suitable for applications in RNA-based therapeutics, biosensing, and synthetic biology.
6. RiboFlow’s performance is further enhanced through a co-design pre-training strategy, which improves geometric awareness by distilling structural priors from RNA crystal structures.
7. Experimental results demonstrate that RiboFlow surpasses baseline models in terms of structural validity, diversity, novelty, and binding affinity, offering a promising tool for ligand-conditioned RNA design.
📜Paper: arxiv.org/abs/2503.17007
#RiboFlow #RNA #RNAEngineering #Bioinformatics #GenerativeModels #DeepLearning #RNASequenceDesign #MoleculeDesign #MachineLearning #AI
4
4
825
1 Jan 2025
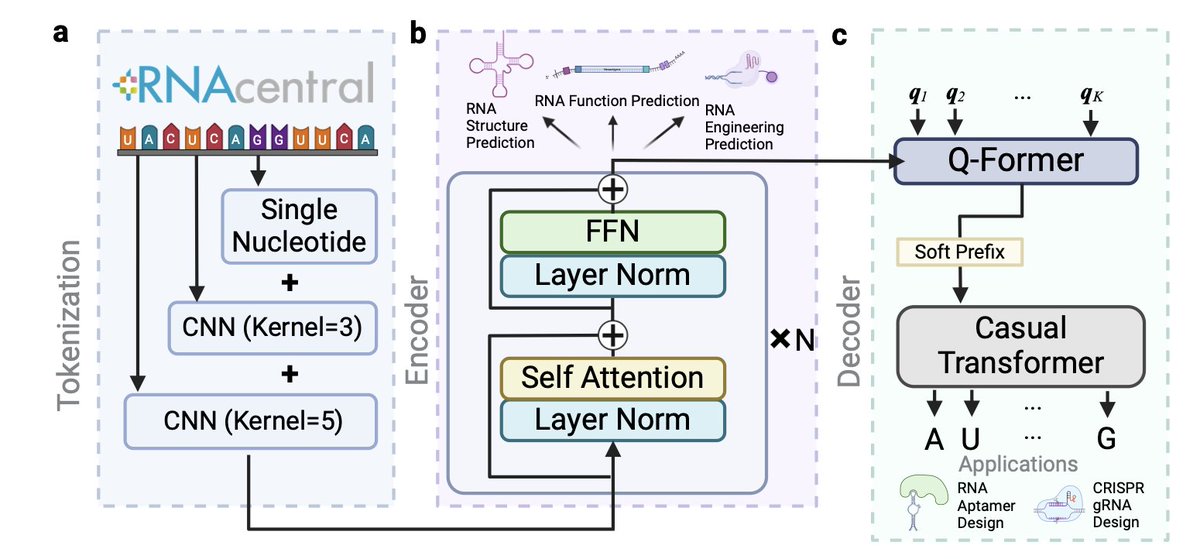
RNAGenesis: Foundation Model for Enhanced RNA Sequence Generation and Structural Insights
1. Introducing RNAGenesis, a novel model bridging RNA sequence understanding and de novo design through latent diffusion. This hybrid system achieves unmatched performance in RNA structure prediction and functional molecule generation.
2. RNAGenesis employs a Bert-like Transformer encoder with Hybrid N-Gram tokenization for rich context understanding, and a Query Transformer to compress latent spaces, paired with an autoregressive decoder for precise sequence generation.
3. The model excels in 9 out of 13 RNA-related tasks, surpassing existing benchmarks like RNA-FM and AIDO.RNA, especially in secondary structure prediction and CRISPR sgRNA optimization.
4. RNAGenesis redefines RNA engineering with its ability to design natural-like RNA aptamers and optimized CRISPR sgRNAs. It offers groundbreaking advancements in biotechnology and RNA therapeutics.
5. Leveraging latent diffusion, RNAGenesis captures RNA sequence distributions with exceptional accuracy, enabling robust modeling of complex RNA motifs and novel molecular designs.
6. Key innovations include the Hybrid N-Gram tokenization scheme and a latent space trained with a denoising diffusion model, ensuring high fidelity in both structure and function predictions.
7. Extensive benchmarks validate RNAGenesis as a state-of-the-art tool, demonstrating superior results in RNA function prediction, vaccine degradation stability, and programmable RNA switches.
8. The model also highlights its robustness in aptamer design, generating sequences closer to natural RNA than competing models, as evidenced by biological metrics like G/C content, secondary structure, and motif clustering.
9. Future directions for RNAGenesis include expanding its capabilities to integrate structural and functional annotations, as well as unifying modeling across RNA, DNA, and protein sequences.
10. RNAGenesis is poised to transform RNA-based research and applications, offering a versatile platform for advancements in gene editing, drug design, and synthetic biology.
@MengdiWang10 @lecong @KaixuanHuang1 @yukang51422
💻Code: github.com/biomap-research/R…
📜Paper: biorxiv.org/content/10.1101/…
#RNA #AI #Bioinformatics #CRISPR #SyntheticBiology #RNAEngineering
14
38
3,437

25 Oct 2023
RNA biosensors have mutation hotspots & incorporating orthogonal base pairs make them faster. #RNAengineering #UnnaturalBasepair #riboswitch #biosensor @ming_hammond @ihirao @sudimanna
pubs.acs.org/doi/10.1021/acs…
3
15
3,448

13 Sep 2021
We had an excellent time at the Central US synbio conference! So many brilliant speakers, panelists, and graduate students. Congratulations to our own August Staubus and Andrea Ameruoso for winning first and second place for their posters! #centralussynbio21 #RNAengineering
1
2
32

13 Nov 2020
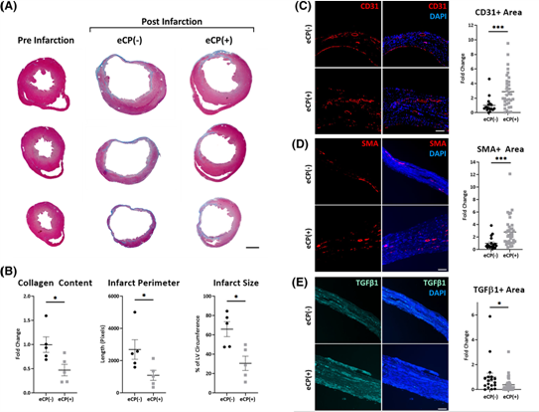
Engineered Cardiopoietic Stem Cell Therapy
A new article reports the engineering of cardiac repair competent stem cells using Brachyury, a mesodermal transcription factor.
#cardiopoiesis #stemcells #heartfailure #regenerativetherapy #RNAengineering
bit.ly/2JzYllh
4
1

15 Mar 2017
15 Mar 2017

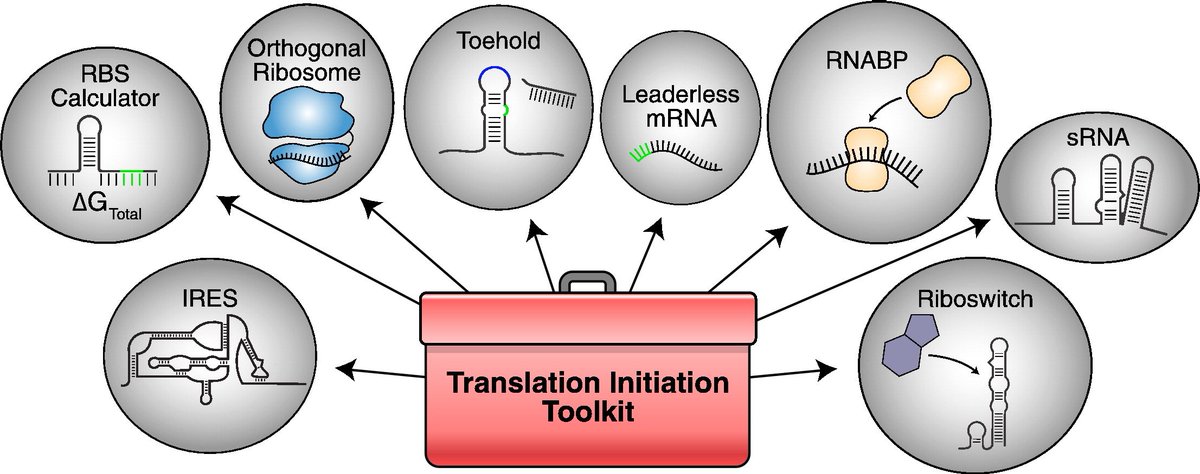
Check out our new review on translation initiation and bioengineering by J. Vigar and Dr. Wieden @uLethbridge sciencedirect.com/science/ar…
2

Thx to Lucks Lab for linking to their Addgene page. Our nonprofit only makes it with your support! hubs.ly/H01Jt1l0 #RNAEngineering
1