An electron-density point-cloud framework for robust protein-ligand interaction prediction
1. E-CloudBind reframes protein–ligand affinity prediction around electron-density point clouds rather than relying on sub-ångström atomic coordinates, aiming to stay accurate when structures are low-resolution or predicted (e.g., AlphaFold) and therefore noisy.
2. Key idea: replace hard distance cutoffs for “contacts” (e.g., within 5 Å) with density-aware Gaussian “electron clouds”, where interactions are defined by overlap/isosurface intersection, yielding a more resolution-agnostic interaction graph.
3. The framework explicitly splits chemistry into two complementary channels: non-covalent interactions from 3D electron-cloud point clouds, and covalent structure from intrinsic molecular graphs (bond topology), then fuses them for affinity regression.
4. Ligand electron density is obtained via semi-empirical quantum chemistry (GFN2-xTB), while protein pockets use a van der Waals radius-guided multivariate Gaussian sampling strategy as a physically motivated proxy that is far cheaper than full QM density.
5. Architecture highlights: K-means clusters point clouds into atom-aligned local regions; a point-cloud encoder (3D-GCN-style deformable kernels) learns local non-covalent patterns (e.g., H-bonds, π-stacking, van der Waals complementarity); a heterogeneous GNN encodes covalent graphs; a multi-bond fusion module integrates both.
6. On PDBbind 10-fold CV, E-CloudBind reports MAE 1.059 and Pearson 0.667, outperforming representative sequence-based (PSICHIC), graph-based (SIGN), and structure-based (DMFF) baselines, and also comparing favorably to recent structure-centric methods (EHIGN, Boltz-2, FlowDock) under the same protocol.
7. Robustness to experimental resolution: when regressing absolute error vs. crystallographic resolution, E-CloudBind shows a much flatter slope (0.017) than baselines (0.053–0.065), with a non-significant trend (p = 0.703), indicating reduced sensitivity to declining structural quality.
8. Robustness to structure source shifts: swapping experimental proteins with AlphaFold2 models causes only a small performance change for E-CloudBind (MAE 0.042; Pearson −0.004), while coordinate-dependent baselines degrade more (e.g., DMFF MAE 0.187; Pearson −0.093).
9. Out-of-distribution testing built from DAVIS via combinatorial partitioning by protein/ligand complexity shows tighter error dispersion for E-CloudBind (lowest median deviation), with stable performance across increasing protein Relative Contact Order and ligand Bertz complexity.
10. Practical and interpretability results: attention maps highlight polar ligand atoms and key pocket regions consistent with known interaction motifs; large-scale screening on 80,383 ZINC molecules against PBP1A, SARS-CoV-2 Mpro, and BCL-2 uses docking for follow-up, plus BCL-2 candidate assessment with synthesizability metrics and explicit-solvent MD (400 ns) suggesting stable binding for selected hits.
💻Code: github.com/Liuyujian0408/DPI ; doi.org/10.5281/zenodo.19851…
📜Paper: doi.org/10.1038/s41467-026-7…
#ComputationalBiology #DrugDiscovery #ProteinLigand #BindingAffinity #GeometricDeepLearning #GNN #PointCloud #ElectronDensity #VirtualScreening #AlphaFold

3
26
1,757
HonestAffinity: Leak-Aware Evaluation of Protein and Pocket Priors for Binding Affinity Prediction
1. HonestAffinity frames a key caution for protein–ligand affinity models: architectural “priors” can flip from helpful to harmful depending on whether evaluation splits leak protein/ligand similarity (canonical CASF/PDBbind-style) or are leak-proof (LP-PDBBind 3-tier no-leak).
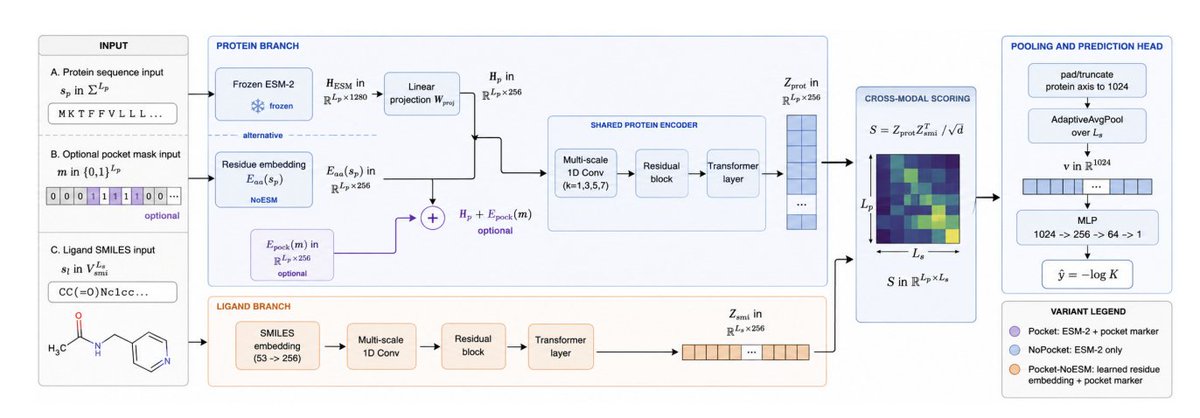
2. The paper isolates two common priors in a controlled 1D-input setting: frozen ESM-2 (650M) per-residue embeddings (1280-d projected to 256) and a learned binary pocket-position marker added to residue features when pocket annotations are available.
3. Core result is a split-conditioned reversal across both priors. On familiar/canonical splits (val, CASF-2016, CASF-2016 non-train), adding ESM-2 and the pocket marker improves performance; on strict LP no-leak tiers (test_cl1–cl3), the same additions reduce Pearson R.
4. Three deployment-matched variants are proposed rather than one “best” model: HONESTAFFINITY-POCKET (ESM-2 pocket marker) for familiar/annotated targets; HONESTAFFINITY-NOPOCKET (ESM-2 only) when no pocket list exists; HONESTAFFINITY-POCKET-NOESM (21-token residue embedding pocket marker) for strict LP-style generalization with pocket annotations.
5. Quantitatively (Pearson R, mean±std over 3 seeds): HONESTAFFINITY-POCKET leads on val (0.548), CASF-2016 (0.747), and CASF non-train (0.646). But on LP strict tiers, HONESTAFFINITY-POCKET-NOESM leads: cl1 0.531, cl2 0.538, cl3 0.497, also giving best RMSE on cl2/cl3.
6. The ESM-2 ablation is especially instructive: swapping ESM-2 for a learned 21-vocab residue embedding decreases R on val/CASF (e.g., CASF-2016 drops from 0.747 to 0.713) but increases R on every strict LP tier (e.g., cl3 rises from 0.433 to 0.497; ∆R up to 0.064).
7. The pocket marker shows the same sign flip: it helps on val/CASF but hurts on LP tiers. Interpretation: both priors inject signals correlated with training-distribution structure (protein-family signatures or pocket geometry), which can become misleading when similarity filtering removes overlap by design.
8. Methodologically, the authors argue for paired reporting: canonical metrics alone would over-credit ESM/pocket priors; leak-proof metrics alone would understate their utility for in-distribution scoring. They recommend routine paired canonical leak-proof ablations for new affinity predictors.
9. Implementation is intentionally compact and scalable: multi-scale 1D CNNs a residual block a single Transformer layer per branch (protein and SMILES), coupled by a matrix-product compatibility map. Training uses 11,513 LP-PDBBind train complexes and runs in ~3 GPU-hours on a single V100; inference is ~10 ms/complex with cached embeddings.
📜Paper: arxiv.org/abs/2606.03422
#CompBio #Bioinformatics #DrugDiscovery #ProteinLanguageModels #ESM2 #BindingAffinity #Benchmarking #PDBbind #CASF2016 #MachineLearning
5
19
1,604
ToolMol: Evolutionary Agentic Framework for Multi-objective Drug Discovery
1. ToolMol addresses a practical failure mode of LLM-based molecule generation: directly emitting SMILES often produces invalid strings (reported >30% invalid even for strong reasoning models), which wastes oracle budget and forces fallbacks to weaker operators in prior work.
2. The key idea is to stop asking the LLM to “write molecules” and instead let it “edit molecules” via tool-calling: an agentic LLM proposes structured modifications, executed deterministically by RDKit-backed functions, making the final output syntactically valid by construction.
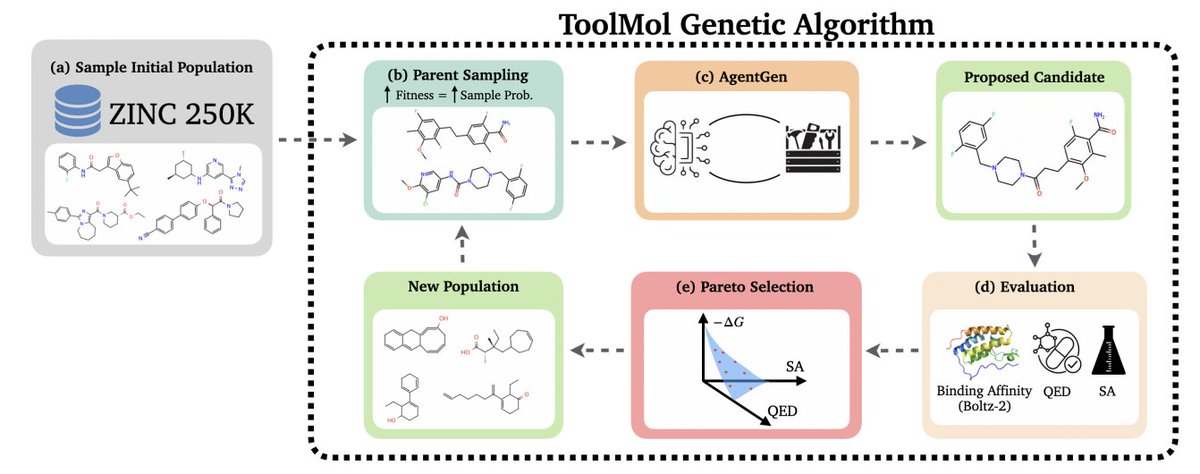
3. Method overview: a multi-objective genetic algorithm maintains a ligand population, selects parents with fitness-proportional sampling, then calls an LLM operator (AgentGen) to perform crossover a few mutations; the next generation is the non-dominated Pareto frontier (binding affinity, QED, SA).
4. The toolbox contains 7 deterministic operations (e.g., crossover molecules, add functional group, replace atom, replace/remove substructure). The LLM supplies parameters (atom indices, groups, bond types), while RDKit enforces valence/graph validity and returns explicit error messages when an operation is impossible.
5. AgentGen is iterative (up to 10 steps; typically fewer). Each tool call updates the conversation with the executed action plus refreshed atom-level structure annotations (substitutable H, ring membership, centrality, etc.) and molecular descriptors (QED, SA, MW, LogP, TPSA, HBD/HBA, rotors).
6. Multi-objective evaluation uses Boltz-2 predicted binding affinity (ΔG), QED (maximize), and SA (minimize). Reporting emphasizes “Filtered Affinity” (top binders that also pass QED > 0.5 and SA < 3.0) plus Pareto hypervolume, reflecting lead-likeness constraints rather than raw affinity alone.
7. Across three targets (c-MET, BRD4, ACAA1), ToolMol ranks best on average and leads on the multi-objective metrics (Filtered Affinity Hypervolume). The paper reports >10% stronger predicted binding affinity than existing methods while producing drug-like and synthesizable candidates.
8. A notable validation step: ToolMol’s top molecules also achieve state-of-the-art Absolute Binding Free Energy (ABFE) results on c-MET and BRD4, improving over MF-LAL by >35% on the reported setup—even though ABFE is not optimized during search (ToolMol optimizes Boltz-2 affinity QED/SA).
9. Ablations isolate why tools matter: swapping ToolMol’s tool-calling operator into MOLLEO’s GA improves results; forcing MOLLEO to retry until valid SMILES reduces invalidity but does not improve (often degrades) optimization metrics, suggesting the gain is not only “validity” but higher-fidelity execution of intended edits.
10. Mechanistic insight from reasoning traces: tool-calling increases concordance between the LLM’s planned chemical changes and the actual applied modifications (ToolMol shows far fewer plan/execution mismatches than direct-SMILES editing), enabling better use of the LLM’s chemical priors during iterative optimization.
📜Paper: arxiv.org/abs/2605.12784
#DrugDiscovery #ComputationalChemistry #Cheminformatics #MolecularDesign #LLM #Agents #ToolCalling #GeneticAlgorithms #MultiObjectiveOptimization #RDKit #ABFE #BindingAffinity
6
37
2,121
Integrating Diffusion and Liquid AI Models for Predicting Peptide Affinity from mRNA Display Selections
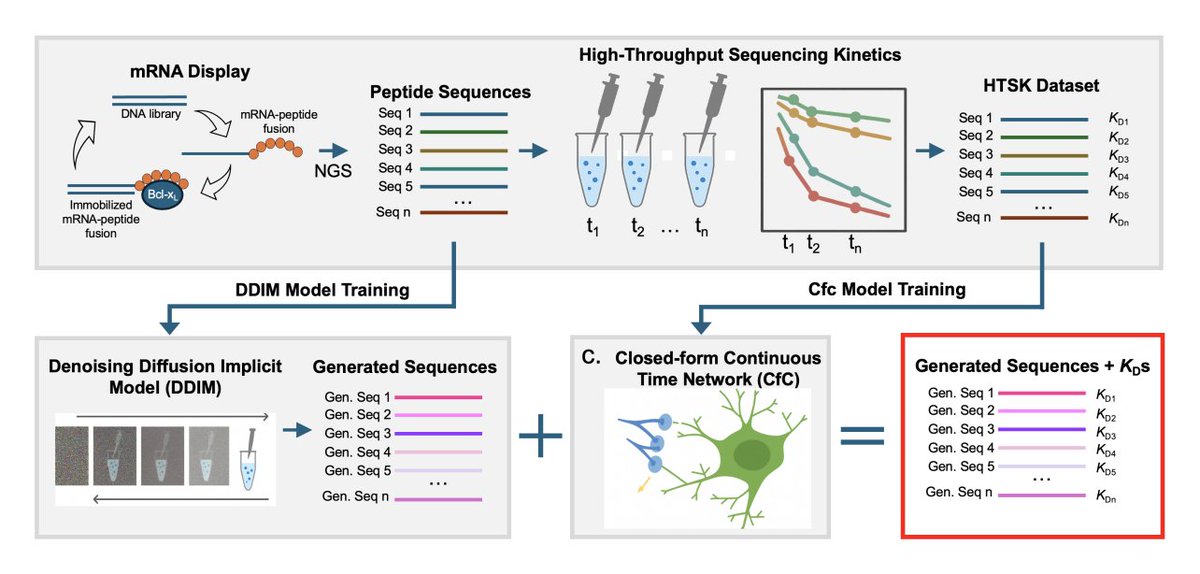
1. The study presents an end-to-end in silico pipeline that couples (i) diffusion-based sequence generation from mRNA display deep sequencing with (ii) a Liquid AI predictor (Closed-form Continuous, CfC) that outputs quantitative binding free energies (ΔG°) for each generated peptide.
2. The biological use case is peptide binding to the oncogenic protein Bcl-xL, leveraging a large, target-specific quantitative dataset from high-throughput sequencing kinetics (HTSK): 15,700 peptides (21-mers) paired with experimentally derived ΔG° values.
3. The key modeling choice is a continuous-time “liquid” neural network (CfC), motivated by the idea that sequence→affinity mapping behaves like an irregular series: amino-acid identity, position, and context produce non-uniform, nonlinear effects, and measured affinities are unevenly distributed across sequence space.
4. CfC is used instead of the earlier Liquid Time-Constant (LTC) networks to avoid expensive numerical solvers: CfC provides a differentiable closed-form dynamics with sigmoid time-gating, reported to yield ~10–100× faster training while retaining accuracy.
5. Model training details: peptides are one-hot encoded as 20×20 matrices; the dataset is split 70:15:15; architecture includes batch norm, dense layers (tanh), two CfC blocks (one returning full sequence, one final state), and a linear output for ΔG°; trained with Adam and MSE for 10 epochs.
6. On held-out HTSK test sequences, predicted ΔG° closely matches the experimental distribution: mean/median both -14.9 kcal/mol; MAE ~0.27 kcal/mol; MSE ~0.12; predictions show mild smoothing and compression mainly at the weaker-affinity end (less represented in training).
7. The diffusion component (DDIM) is trained on selection-derived sequencing data to generate novel functional peptides beyond the experimentally observed pool; 20,000 new sequences are sampled with near-identical positional amino-acid frequency to the training distribution (KL divergence 0.0036).
8. The combined workflow (DDIM generation CfC scoring) is experimentally validated: multiple DDIM-generated candidates predicted to be strong binders are synthesized as mRNA–peptide fusions and kinetically measured; several show single-digit picomolar affinities, including ~4-fold improvements over the E1 reference peptide.
9. Quantitatively, across 14 kinetically evaluated DDIM-generated peptides (DDIM-HTSK1–4 and FG variants), CfC predictions agree well with experiments (reported MSE < 0.4 kcal/mol); importantly, the model can rank close sequence neighbors differently when affinity differs substantially (e.g., FG7 vs DDIM-HTSK4).
10. Limitations are clearly demonstrated: when DDIM proposes mutations in an extremely conserved motif region (e.g., positions 10–15 “Y-KAAD” where 99.9% of training sequences match), CfC may still predict high affinity, yet pull-down experiments show little binding—highlighting out-of-distribution and low-sampling failures, and the need for broader dynamic range and coverage.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #ProteinEngineering #PeptideDesign #mRNADisplay #DirectedEvolution #MachineLearning #DiffusionModels #LiquidNeuralNetworks #BindingAffinity #BclxL
7
16
1,557
Pan–Pharmacological Drug–Target Interaction Prediction with 3D–Informed Protein Encoding at Scale
1 OmniBind is a multitask DTI framework that predicts four pharmacological endpoints at once (pKd, pKi, pIC50, pEC50) from a single compound–protein pair, aiming to output a “pan-pharmacological profile” rather than optimizing for only one assay type.
2 The key scaling idea is to bring protein 3D information into sequence-speed inference: protein tertiary structure is converted into a 1D 3Di token sequence (20-letter structural alphabet) generated from sequence-derived structure, then modeled with Transformers like a language sequence.
3 Proteins are encoded in two parallel modalities: (a) amino-acid sequence Transformer encoder (captures evolutionary/sequence cues) and (b) 3Di-structure Transformer encoder (captures local biophysical environments). A learnable gated fusion layer dynamically weights each modality per input, instead of static addition/concatenation.
4 On the compound side, OmniBind uses a molecular graph representation from SMILES with a 1-layer GCN (chosen to avoid over-smoothing). A “virtual atom” connected to all atoms is added to aggregate global context, following prior CPI Transformer designs.
5 Compound–protein interaction is modeled by a 5-layer Transformer decoder: self-attention over compound atoms plus cross-attention to the fused protein representation. The virtual-atom embedding is used to emit four endpoint predictions in parallel.
6 Scale and data realism: trained on BindingDB May 2023 after extensive cleaning and unit harmonization (all endpoints converted to pActivity = −log10(M)). The final training snapshot contains 2,282,997 compound–protein pairs; temporal benchmarking uses newly added records in BindingDB Nov 2024.
7 Ablations show why gating matters: sequence-only and 3Di-only perform similarly; naive combination (element-wise addition) fails to consistently improve; gated fusion is best across regression and classification metrics, supporting the claim that sequence and 3Di provide complementary signals that must be context-weighted.
8 Generalization is tested with two protocols designed to penalize shortcut learning: (a) label reversal test (adversarial ligand-bias probe) and (b) temporal validation where both compounds and proteins are unseen (train up to May 2023; test on new Nov 2024 entries). OmniBind outperforms DTI-LM and TransformerCPI2.0 in both settings, suggesting it relies less on ligand–protein co-occurrence patterns.
9 Interpretability: cross-attention analysis on ABL1–imatinib highlights the known gatekeeper residue T315 (mapped to residue 93 in the kinase-domain construct). Introducing the resistance mutation T315I reduces attention at that position, indicating sensitivity to clinically relevant single-residue changes.
10 Practical demonstrations: proteome-wide screening against 20,421 reviewed human proteins recovers clozapine’s known clinical target landscape strongly (6/7 known therapeutic/off-targets within top 200; HTR2A ranked #1), and distinguishes clozapine vs clomipramine despite shared tricyclic scaffolds (HTR2A vs SLC6A4 as top-ranked primary targets). A separate repositioning workflow screens 1,615 FDA-approved drugs against PDE5, KLKB1, SIRT3 and uses Boltz-2 MOE docking for structural plausibility checks, yielding avanafil (PDE5) as an external positive control and proposing glecaprevir (KLKB1) and valrubicin (SIRT3) as candidates.
📜Paper: biorxiv.org/content/10.64898…
#ComputationalBiology #Bioinformatics #DrugDiscovery #DTI #DeepLearning #Transformers #ProteinStructure #MultitaskLearning #BindingAffinity #Cheminformatics

2
23
1,523
On the rise of AI technologies in virtual screening
1. Cecchini & Sinenka benchmark Boltz-2 on ULVSH, a notoriously hard virtual-screening rescoring dataset built from 10 ultra-large docking hit lists followed by in vitro binding assays (943 compounds total; 427 actives, 516 inactives; targets include 7 GPCRs plus ROCK1, a membrane receptor, and a transporter).
2. Core result: Boltz-2 is the top-performing classifier among all rescoring strategies compared (empirical scores, ML rescoring, single-point energy methods incl. polarizable FF / semi-empirical QM, and end-point free-energy approaches), reaching mean ROC-AUC 0.70 across targets.
3. Using a practical criterion from prior work (ROC-AUC > 0.7 as “successful”), Boltz-2 succeeds on 7/10 targets, more than doubling the best success rate among the alternative rescoring methods tested previously on the same benchmark.
4. Practical workflow: starting only from protein FASTA sequences (from PDB) and ligand structures (MOL2), they implement an automated pipeline to generate Boltz-2 inputs, run co-folding affinity prediction, and collect ligand rankings using Boltz-2’s affinity_probability_binary (with similar results using affinity_pred_value).
5. Throughput on commodity hardware: ~100 seconds per ligand on a single GPU (reported on RTX 4500 Ada and RTX A4500), enabling the full ULVSH set in ~1 day; results are reproducible across GPUs with per-target ROC-AUC deviations < 0.04.
6. Failure analysis: two targets (CNR1 and MTR1A) are clear misses (ROC-AUC ~0.40–0.45). The authors test multiple mitigation hypotheses—ensembling (20 repeats/ligand), template conditioning with experimental structures, restricting to single domains, higher-quality inference settings, and more stochastic sampling—but none improves over defaults in a consistent way.
7. Structural insight: Boltz-2 places ligands in the correct binding pocket relative to known co-crystals, but pose RMSD is >2 Å in 5/10 cases. Notably, pose accuracy does not correlate with classification performance, suggesting the affinity signal can be decoupled from geometric fidelity in this setting.
8. More “realistic” hit-identification test using LSD docking lists (20M–468M compounds per target): for 6 targets, they take the top-1,000 docking hits and spike in all known actives (including those ranked low by docking). Re-ranking by Boltz-2 yields ~4–5x enrichment over docking alone across top-100 to top-750 selections (target-dependent; some N/A where data not available).
9. Positioning: Boltz-2 is far slower than docking (orders of magnitude), so it is framed as a rescoring layer rather than a docking replacement—potentially bridging ultra-large docking (~10^9) to tractable experimental/testing sets (~10^3–10^5), and possibly seeding downstream ABFE with Boltz-2 poses.
10. Context and caveats: the manuscript notes contemporaneous studies broadly agreeing that co-folding can improve classification vs conventional docking, while raising concerns about domain of applicability (generalization beyond training distribution, sensitivity to mutations, and potential ligand-dominant behavior in extreme cases). The authors conclude prospective validation is still needed.
📜Paper: biorxiv.org/content/10.64898…
#VirtualScreening #DrugDiscovery #ComputationalChemistry #CADD #ProteinLigand #BindingAffinity #AI4Science #GPCR #MachineLearning #Boltz2

1
2
29
2,520
Effects of Protein Interface Mutations on Protein Quality and Affinity
1. The study argues that many high-throughput “affinity” measurements actually mix two signals: true interface energetics (protein-interaction) and biophysical viability (protein-quality: folding/stability/expression). This confounding can mislead both model training and benchmarking.
2. They introduce an experimental analytical framework to disentangle these components in antibody–antigen binding by using control binders: a control VHH binds the same antigen but a non-overlapping epitope, so epitope mutations should not directly change that interaction unless antigen protein-quality changes.
3. Dataset scale: AlphaSeq yeast-display mating DMS across four VHH–antigen complexes (SARS-CoV-2 RBD and Botulinum neurotoxin A), covering 7,185 mutations total, including single and double mutants in both antigen epitopes and VHH paratopes (with multiple synonymous WT replicates to denoise oKD estimates).
4. Key quantitative result for antigen epitope mutations: most observed affinity losses are dominated by antigen protein-quality deterioration rather than interface disruption. Among mutations that substantially hurt primary binding (ΔoKD > 1), 43%–82% of singles and 42%–83% of doubles had ΔΔoKD < 1 (primary vs control shift differs by <10-fold), consistent with “mostly protein-quality” rather than “mostly protein-interaction”.
5. They validate that the control VHH readout is largely tracking global antigen quality (not idiosyncratic allostery) by showing very high agreement among multiple additional controls (average Spearman 0.96 across 6 additional controls), implying many mutations reduce functional antigen abundance broadly.
6. Structural/biophysical mapping: positions classified as “protein-interaction” (top 20% by median ΔΔoKD) tend to be residues making dense interfacial contacts—especially hydrogen bonds, polar and ionic interactions—whereas “protein-quality” positions often make few/no direct interfacial bonds and show more conservative substitutions.
7. A consistent (but modest) biochemical trend is that protein-interaction effects are more associated with charge/polarity changes than protein-quality effects; hydropathy/volume signals appear more context-dependent across complexes.
8. Model benchmarking on disentangled labels shows a major implication: several popular methods correlate with observed affinity largely because they capture protein-quality, not interface energetics. ThermoMPNN and ESM-IF1 show the strongest correlations to ΔoKD for antigen mutants, but they predict control-binder ΔoKD nearly as well as primary-binder ΔoKD—evidence the dominant learned signal is protein-quality.
9. For the subset of mutations enriched for protein-interaction effects, model performance drops for ThermoMPNN and ESM-IF1 single-chain; ESM-IF1 multi-chain helps somewhat, but overall interface-specific prediction remains limited. For double mutants, ESM-IF1 performs worse than for singles and struggles particularly with non-additive (epistatic) effects.
10. Antibody-side mutations are harder: across VHH paratope mutations (mostly CDR loops), existing models show lower correlations to ΔoKD than for antigen mutations, consistent with the idea that antigen scans are dominated by protein-quality (easier for models), while paratope scans contain more true interaction signal (harder for models).
📜Paper: biorxiv.org/content/10.64898…
#ProteinEngineering #Antibodies #DeepMutationalScanning #ProteinDesign #BindingAffinity #ProteinStability #ComputationalBiology #MachineLearning #StructuralBiology #YeastDisplay

5
29
2,122
Single-Pass Discrete Diffusion Predicts High‑Affinity Peptide Binders at >1,000 Sequences per Second across 150 Receptor Targets
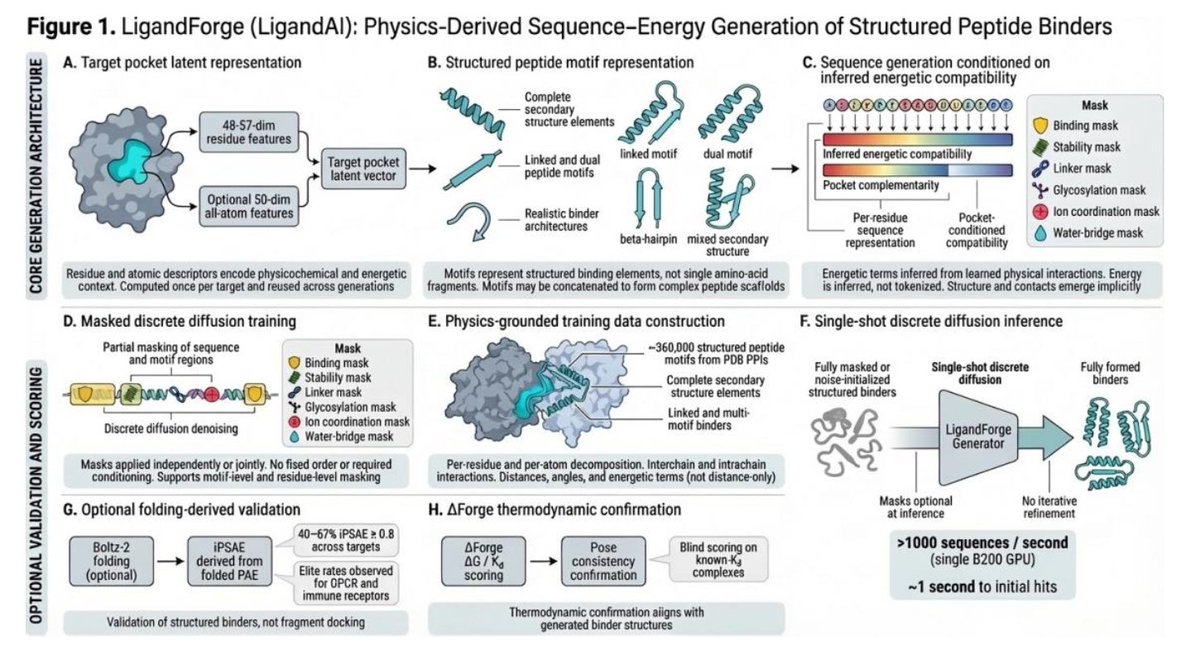
1. The authors introduce LigandForge, a discrete diffusion model that generates peptide sequences directly from a receptor pocket’s 3‑D geometry—no intermediate structure prediction or inverse folding step is required.
2. By embedding explicit thermodynamic supervision (hydrogen‑bond energies, van der Waals contacts, ΔG, and per‑residue interaction penalties) into the training objective, LigandForge internalizes binding physics, eliminating the need for costly structure refinement at inference.
3. Throughput is unprecedented: LigandForge produces ~700–1,000 sequences per second on a single GPU, a 10,000‑fold speed‑up over BoltzGen and a million‑fold advantage over BindCraft, enabling whole‑target portfolio screening in minutes.
4. In a five‑target benchmark (TNF‑α, PD‑L1, VEGF‑A, IL‑7Rα, HER2), LigandForge generated 150,000 candidates in 3.4 min and identified predicted sub‑100 nM binders for all five, whereas BoltzGen found only two and BindCraft none.
5. Structural validation with Boltz‑2 and thermodynamic scoring with the Rust‑based DeltaForge (r = 0.83 on a high‑quality peptide benchmark) revealed that 73 % of targets had sub‑100 nM predicted binders and 30 % had sub‑1 nM, across diverse protein classes including GPCRs, transporters, and homodimers.
6. LigandForge’s sequence‑space diffusion produces markedly diverse designs (mean pairwise identity ≈ 4 %) and a broader secondary‑structure palette (≈ 70 % helices, 9 % β‑sheets, 10 % coil) compared to backbone‑sampling methods, which are heavily helix‑biased.
7. The platform natively supports multimeric receptors, enabling bivalent engagement in KIT homodimers and orthosteric pocket embedding in aminergic GPCRs (DRD2, HTR2A) without any tailoring of the model or pipeline.
📜Paper: biorxiv.org/content/10.64898…
#PeptideDesign #DiffusionModels #ComputationalBiology #ProteinEngineering #GPCR #MachineLearning #BindingAffinity #Thermodynamics
8
49
3,191
AbAffinity: A Large Language Model for Predicting Antibody Binding Affinity against SARS-CoV-2
1 AbAffinity achieves state-of-the-art performance in predicting antibody binding affinity against SARS-CoV-2, with Pearson correlation of 0.655 and Spearman correlation of 0.608, significantly outperforming existing methods including DG-Affinity, ESM-2, and AbLang.
2 The model is built on a fine-tuned ESM-2 architecture with 33 layers and 650M parameters, specifically trained on 71,834 unique scFv antibody sequences with experimentally measured binding affinities against a conserved HR2 peptide region of SARS-CoV-2.
3 Unlike generic protein language models, AbAffinity learns SARS-CoV-2-specific binding patterns through task-specific fine-tuning, enabling it to capture mutation effects and create meaningful embedding spaces that correlate smoothly with binding affinity.
4 The model generates interpretable residue-residue attention maps that highlight complementarity-determining regions (CDRs) as critical for binding prediction, providing biological insights into strong versus weak binding antibodies.
5 AbAffinity's embeddings demonstrate unexpected versatility by capturing thermostability properties of antibodies without explicit training on this attribute, and enable superior performance on downstream classification tasks compared to baseline embeddings.
6 The model is publicly available through PyPI, allowing seamless integration into antibody design workflows for therapeutic development against COVID-19 and potentially other infectious diseases.
💻Code: pypi.org/project/AbAffinity/
📜Paper: arxiv.org/abs/2603.04480
#AbAffinity #AntibodyDesign #SARSCoV2 #BindingAffinity #ProteinLanguageModel #ComputationalBiology #AIforScience #TherapeuticAntibodies

6
22
1,786
On Improving Experimental Binding Affinity Predictions with Synthetic Data
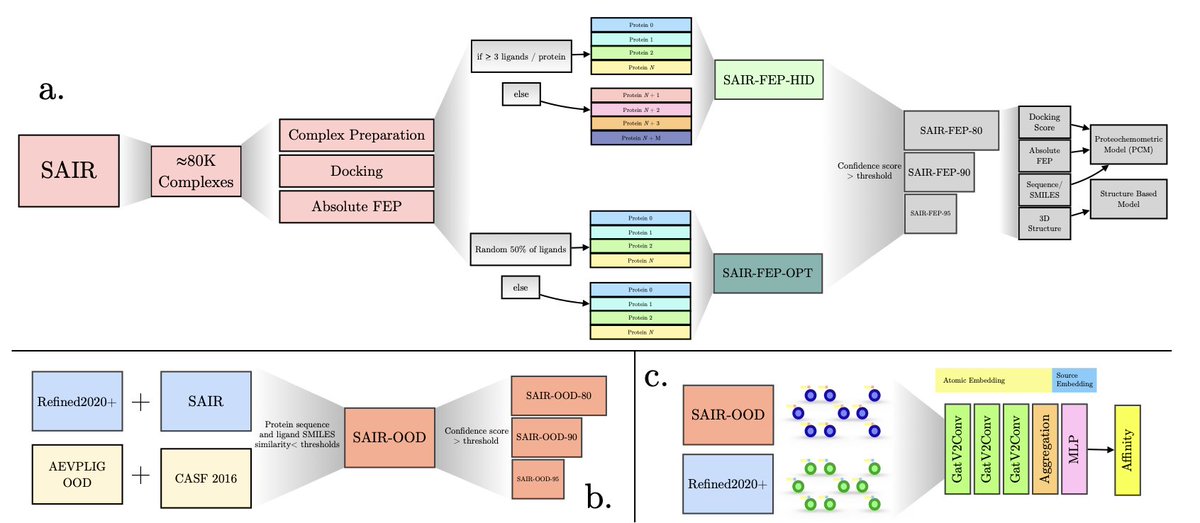
1. This study from SandboxAQ presents a comprehensive analysis of how synthetic structural data can enhance deep learning models for predicting protein-ligand binding affinities, addressing a critical bottleneck in computational drug discovery.
2. The authors generated approximately 80,000 absolute free energy perturbation (AFEP) calculations and created two specialized datasets: SAIR-FEP for benchmarking different drug discovery scenarios and SAIR-OOD for rigorous out-of-distribution testing.
3. A key finding reveals that structure-based deep learning models are highly sensitive to input structure quality—filtering for high-confidence co-folded complexes from Boltz-1x leads to predictable performance improvements, while blindly adding lower-quality structures causes metrics to saturate.
4. The research demonstrates that proteochemometric (PCM) models, which use sequence and SMILES-based representations, can be significantly enhanced by incorporating physics-based descriptors like docking scores and free energy values.
5. Most importantly, simultaneous training on both synthetic and experimental data consistently improves model performance on established experimental benchmarks, with up to 8% reduction in RMSE and 9-11% increases in correlation metrics on CASF 2016.
6. The study recovers traditional neural scaling laws only when using high-quality synthetic structures, suggesting that careful data curation is essential for realizing the benefits of large-scale computational data augmentation in affinity prediction.
7. These results provide clear practical guidance for integrating synthetic structural data into drug discovery workflows, emphasizing quality over quantity when augmenting experimental datasets.
📜Paper: biorxiv.org/content/10.64898…
#DrugDiscovery #MachineLearning #ComputationalChemistry #BindingAffinity #FreeEnergyPerturbation #DeepLearning #Cheminformatics #Bioinformatics #ProteinLigand #AIDD
1
1
22
2,144
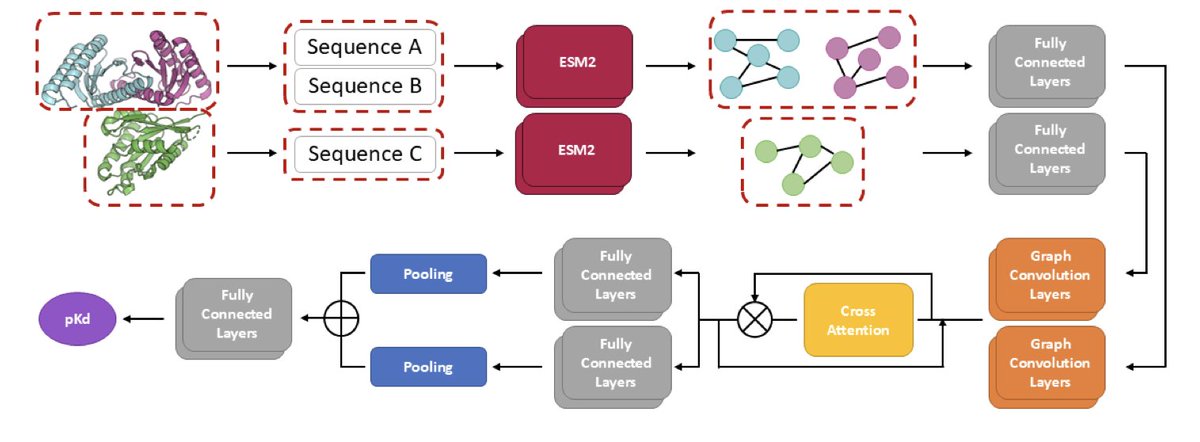
CrossAnity: A Sequence-Based Protein-Protein Binding Affinity Prediction Tool Using Cross-Attention Mechanism
1 CrossAnity achieves state-of-the-art performance among sequence-based models and even outperforms structure-based models on the challenging S34 test set, which contains newer protein complex structures that the model never encountered during training.
2 The model introduces a novel cross-attention mechanism that enables two interacting protein components to exchange contextual information, allowing the model to capture interaction patterns without requiring expensive 3D structural data.
3 By leveraging ESM2 embeddings combined with graph convolutional networks built from predicted contact maps, CrossAnity extracts rich evolutionary and spatial information directly from amino acid sequences.
4 The architecture demonstrates remarkable inference speed, completing predictions in approximately 20-56 seconds on GPU compared to hundreds or thousands of seconds required by structure-based competitors.
5 An ablation study confirms that ESM2 transfer learning provides the most critical information, while cross-attention and graph convolutional layers contribute substantially to prediction accuracy.
6 Two case studies validate practical utility: alanine-scanning mutagenesis reveals the model's ability to identify key interface residues, and nanobody ranking against SlyD contaminant shows 0.776 Pearson correlation with experimental binding affinities.
7 As a sequence-only approach, CrossAnity addresses a critical limitation in drug discovery where most protein complex structures remain undetermined, enabling rapid screening of large peptide libraries for therapeutic development.
💻Code: github.com/guanjsj/CrossAnit…
📜Paper: biorxiv.org/content/10.64898…
#ProteinProteinInteraction #BindingAffinity #DeepLearning #DrugDiscovery #PeptideDesign #ESM2 #CrossAttention #ComputationalBiology #Bioinformatics #AIforScience
2
26
2,225
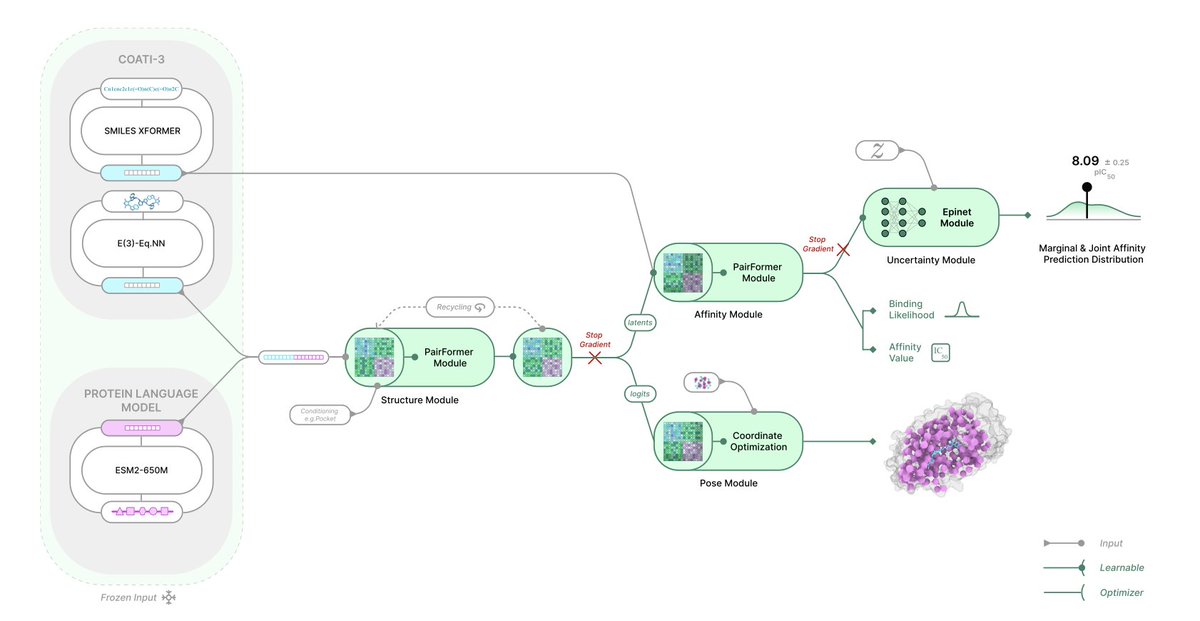
TerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations
1. TerraBind achieves 26-fold faster inference than state-of-the-art methods like Boltz-2 while improving binding affinity prediction accuracy by approximately 20%, addressing a critical computational bottleneck in structure-based drug design.
2. The core innovation challenges the prevailing assumption that full all-atom diffusion is necessary for accurate predictions. Instead, TerraBind uses a coarse pocket-level representation with only protein Cβ atoms and ligand heavy atoms, eliminating expensive generative modeling.
3. The architecture combines frozen pretrained encoders—COATI-3 for molecular representations and ESM-2 for protein sequences—with a lean 48-layer pairformer trunk of just 27M parameters, compared to Boltz-2's 509M parameters.
4. For pose generation, TerraBind employs a diffusion-free optimization module that produces 3D coordinates in under 0.2 seconds, matching diffusion-based baselines on FoldBench, PoseBusters, and Runs N'Poses benchmarks.
5. The binding affinity module operates directly on structural pairformer representations without requiring coordinate generation, outperforming Boltz-2 on 15 of 18 proprietary drug discovery targets and achieving superior Pearson correlation on CASP16.
6. A built-in uncertainty quantification system uses pairwise distance entropy as a zero-shot confidence metric, validated to correlate with both pose accuracy and binding strength without separate training.
7. The epistemic neural network (epinet) module provides calibrated affinity uncertainty estimates, enabling a continual learning framework that achieves 6× greater affinity improvement over greedy selection strategies in simulated drug discovery cycles.
8. Structural fine-tuning on minimal proprietary crystallographic data (as few as 3-6 structures) yields 17% affinity improvement on held-out compounds, demonstrating practical adaptability for specific drug programs.
📜Paper: arxiv.org/abs/2602.07735
#TerraBind #DrugDiscovery #MachineLearning #ProteinLigand #BindingAffinity #StructurePrediction #ComputationalBiology #AIforScience
1
13
47
2,854

9 Dec 2025
Learning to Generalize: Deep Models and Robust Benchmarks for Drug–Target Affinity Prediction
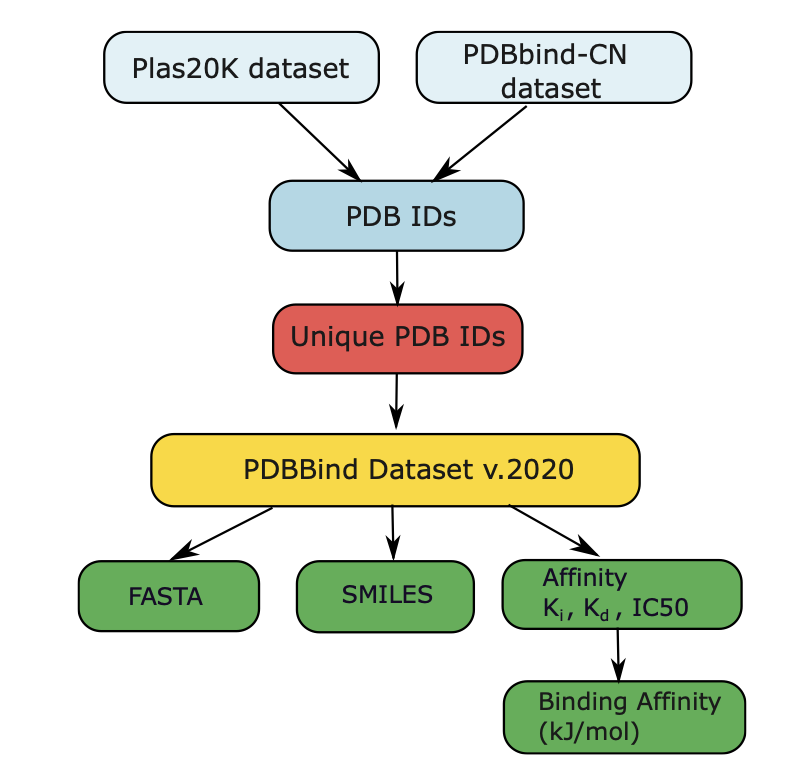
1. This study introduces two novel sequence-based models, AffinityLM and AffinityNet, designed to predict drug–target binding affinity without relying on 3D structures. These models leverage FASTA and SMILES sequences to capture biochemical interactions, offering a scalable alternative to traditional methods.
2. The authors curated a new dataset of approximately 14,002 protein–ligand pairs, each mapped to a unique PDB ID and associated with experimentally determined binding affinities. This dataset addresses data leakage issues by ensuring no overlap of proteins and ligands across training, validation, and test sets.
3. AffinityNet, an attention-based architecture, integrates graph attention for local context and transformer blocks for long-range dependencies. It demonstrates superior performance, achieving a Pearson correlation coefficient of 0.67 and a mean absolute error of 5.8 kJ/mol, outperforming existing benchmarks.
4. AffinityLM utilizes embeddings from pre-trained biological–chemical language models (ESM-2 and MolFormer) to generate feature representations. While competitive, it shows limitations in generalizing to out-of-distribution datasets compared to AffinityNet.
5. The study highlights the importance of rigorous data splitting strategies, showing that traditional random splits can inflate model performance metrics by over 20% compared to more realistic cluster-based evaluations.
6. Independent tests on a molecular docking-based dataset and the CSAR-HiQ_36 dataset further validate AffinityNet's robust generalization capabilities, achieving correlations of 0.65 and 0.78 respectively, significantly outperforming other state-of-the-art models.
📜Paper: doi.org/10.26434/chemrxiv-20…
#DrugDiscovery #DeepLearning #Bioinformatics #ProteinLigand #BindingAffinity #AIinBiology
1
9
1,406
1 Dec 2025
Beyond Atoms: Evaluating Electron Density Representation for 3D Molecular Learning
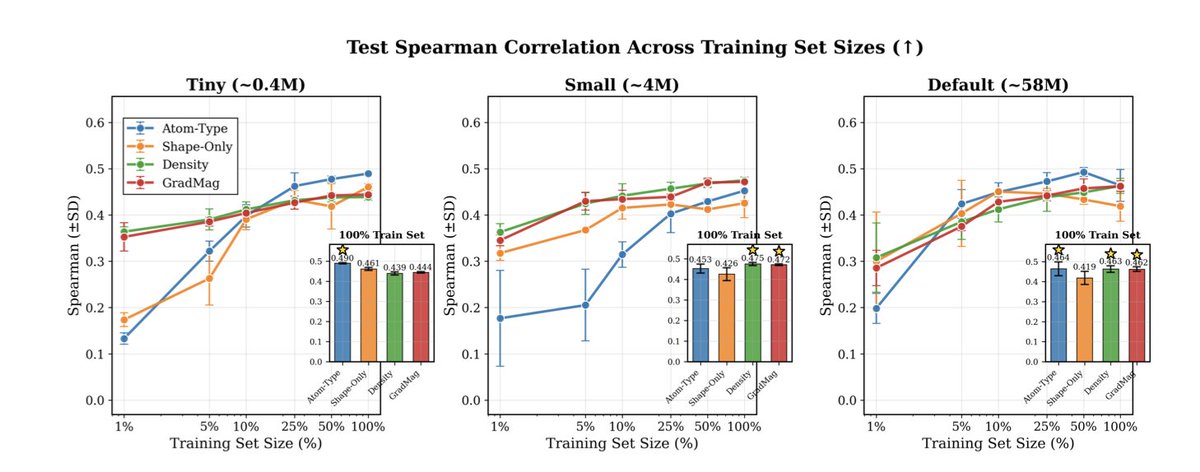
1. A new study explores the potential of electron density maps as inputs for 3D convolutional neural networks (CNNs) in molecular property prediction, challenging the traditional atom-based representations. The research compares atom types, raw electron density, and density gradient magnitude across two tasks: protein–ligand binding affinity prediction and quantum property prediction.
2. In the low-data regime for protein–ligand binding affinity prediction, electron density-based inputs outperform atom-type representations, highlighting the importance of spatial occupancy and continuous electron distribution in capturing binding interactions. This suggests that density maps may offer a richer signal for learning molecular properties governed by electronic structure.
3. For quantum property prediction, density-based inputs consistently outperform atom-based ones at full data scale, despite using less accurate semiempirical methods to generate the input densities. This indicates that electron density captures essential aspects of electronic structure not easily recovered from atom types alone.
4. The study demonstrates that the effectiveness of different molecular representations is task-dependent. While atom-type information is crucial for quantum property prediction, spatial occupancy alone can be highly predictive for binding affinity in certain datasets. This insight could guide the selection of representations based on the specific requirements of molecular prediction tasks.
5. The research highlights the potential of electron density maps as a physically grounded alternative to atom-based representations in 3D molecular learning. It suggests that density-derived inputs can improve data efficiency and accuracy in certain scenarios, paving the way for new approaches in structure-based drug discovery and molecular property prediction.
📜Paper: arxiv.org/abs/2511.21900v1
#MolecularLearning #ElectronDensity #3DCNNs #QuantumProperties #BindingAffinity #MachineLearning
6
946
21 Oct 2025
From Water Networks to Binding Affinities: Resolving Solvation Dynamics for Accurate Protein-Ligand Predictions
1. This study presents a novel approach to accurately predict protein-ligand binding affinities by resolving the complex dynamics of water networks using the polarizable AMOEBA force field combined with Lambda-ABF-OPES. This method effectively captures the dynamic role of water molecules in binding sites, which is crucial for reliable binding free energy estimation.
2. The Lambda-ABF-OPES framework integrates lambda-dynamics with multiple-walker adaptive biasing force and on-the-fly probability enhanced sampling. This combination allows for efficient rehydration sampling of the binding site without explicitly including water-related collective variables, overcoming challenges in sampling water reorganization and protein conformational changes.
3. The study demonstrates the method's effectiveness across diverse protein-ligand complexes, including those with buried and semi-buried ligands. The results show good agreement with experimental data, highlighting the robustness and reproducibility of the approach in capturing dynamic water networks and providing accurate binding free energy estimates.
4. The use of the polarizable AMOEBA force field is a key innovation, as it allows for a more realistic description of solvation and protein-ligand interactions compared to classical fixed-charge force fields. This leads to improved accuracy in modeling hydration effects and water-mediated interactions.
5. The study also provides detailed insights into the hydration dynamics of specific complexes, such as the TAF1(2) bromodomain and the major urinary protein 1. These analyses reveal the nuanced interplay between water mobility and binding affinity, showcasing the method's ability to provide mechanistic understanding of molecular interactions.
📜Paper: biorxiv.org/content/10.1101/…
#ProteinLigandBinding #SolvationDynamics #MolecularDynamics #BindingAffinity #ComputationalBiology

11
31
2,287
4 Oct 2025
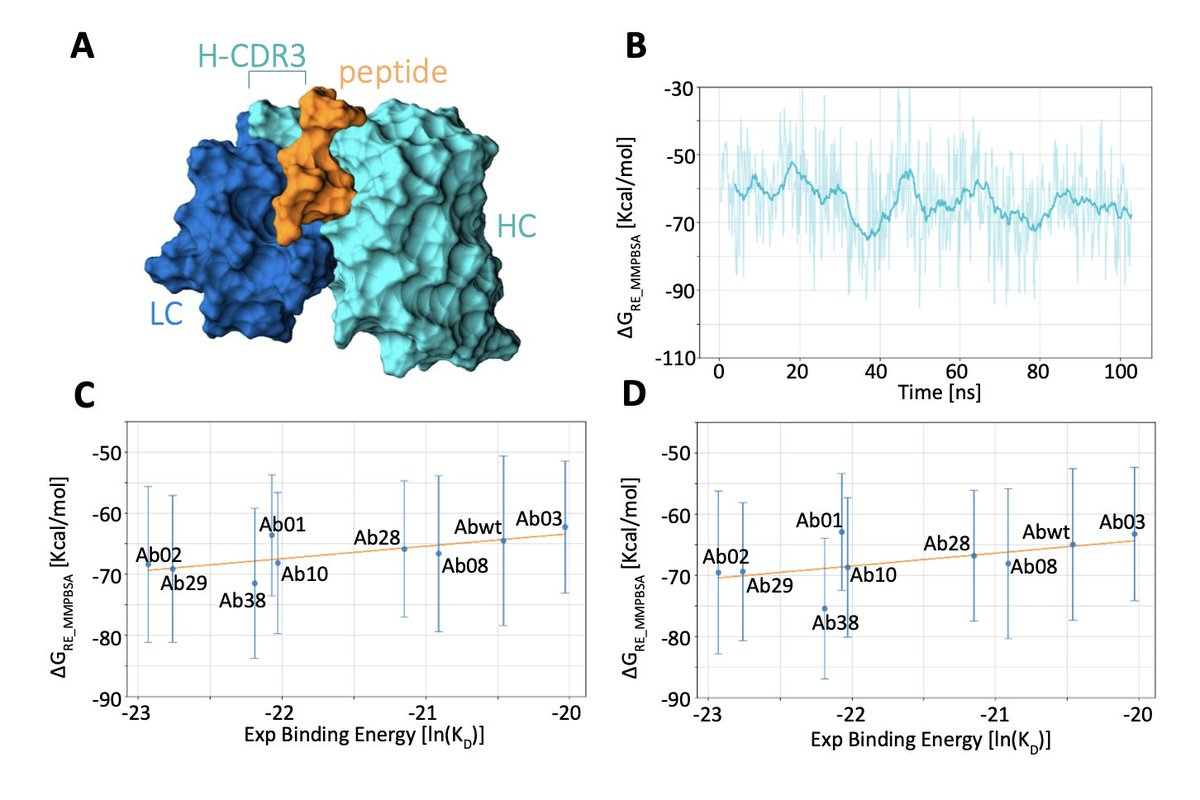
Assessing computational strategies for the evaluation of antibody binding
1. This study provides a comprehensive comparison of different computational methods for calculating antibody binding affinities, highlighting the importance of accurate binding affinity prediction in drug discovery. The research explores various strategies, including MMPBSA, replica exchange molecular dynamics, and potential of mean force calculations, to identify the most effective approaches for evaluating antibody-peptide interactions.
2. The study finds that equilibrium MMPBSA calculations offer better agreement with experimental binding affinities than non-equilibrium potential of mean force evaluations, emphasizing the system-dependent nature of these methods. This finding challenges some previous reports and underscores the need for careful selection of computational techniques based on the specific characteristics of the system under study.
3. The research also investigates the impact of enhanced sampling techniques, such as replica exchange molecular dynamics, on the accuracy of binding affinity predictions. While this method improves accuracy, it comes at a significantly higher computational cost. The study suggests that careful calibration of simulation length is crucial for achieving a balance between accuracy and computational feasibility.
4. Interestingly, the study observes that longer simulations do not always lead to more accurate predictions, with shorter simulations sometimes yielding better results. This counterintuitive finding highlights the complexities involved in molecular dynamics simulations and suggests that optimal simulation length may need to be empirically determined for each system.
5. The study concludes that computational methods based on molecular dynamics simulations, when paired with extensive sampling of the configuration space, provide a valuable tool for antibody screening. The simpler protocol of running multiple short simulation replicas emerges as an effective compromise between accuracy and computational feasibility, offering a practical approach for rapid screening in drug design pipelines.
📜Paper: biorxiv.org/content/10.1101/…
#ComputationalBiology #DrugDiscovery #AntibodyBinding #MolecularDynamics #Simulation #BindingAffinity #MMPBSA #ReplicaExchange #PotentialOfMeanForce
12
62
3,466
30 Sep 2025
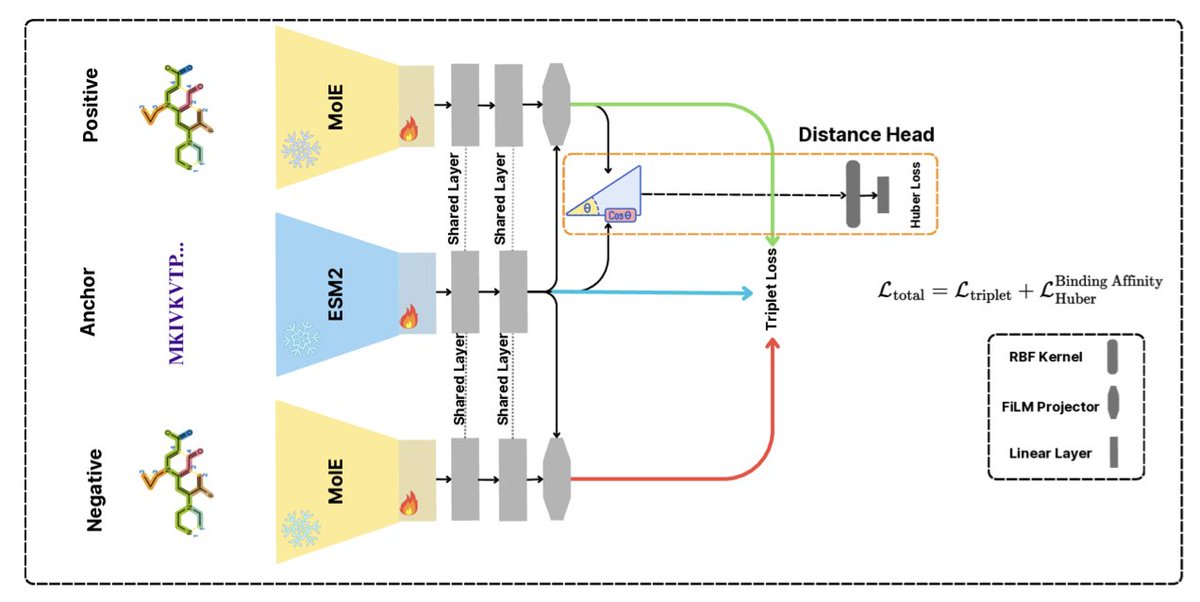
Learning to Align Molecules and Proteins: A Geometry-Aware Approach to Binding Affinity
1. A novel study introduces FIRM-DTI, a lightweight yet powerful framework for predicting drug–target binding affinity, achieving state-of-the-art performance with fewer parameters than large models. This innovative approach leverages a FiLM conditioning layer and triplet loss to enhance generalization across chemical space and time, addressing limitations of previous methods that often rely on simple concatenation of features.
2. The core novelty of FIRM-DTI lies in its geometry-aware design. By using a FiLM layer, the model can modulate molecular embeddings based on protein embeddings, allowing for target-specific transformations. This is complemented by a triplet metric-learning objective that pulls interacting pairs closer and pushes non-interacting pairs apart in the embedding space, resulting in more robust and interpretable predictions.
3. The study demonstrates the effectiveness of FIRM-DTI through extensive experiments on the Therapeutics Data Commons DTI-DG benchmark and other standard datasets. Despite its modest size, FIRM-DTI outperforms recent large models and achieves a Pearson correlation coefficient of 0.59 on the DTI-DG test set, surpassing state-of-the-art sequence-based and network-based approaches.
4. The authors highlight the importance of metric learning by showing a significant performance drop when the triplet loss is omitted. This underscores the value of enforcing geometric structure in the latent space for better generalization. The learned RBF head provides a smooth mapping from cosine distance to binding affinity, further confirming the model's geometry-awareness.
5. The research also explores drug–target interaction classification, achieving high AUPR and AUROC scores on BIOSNAP and BindingDB datasets. This indicates that the FiLM-conditioned, geometry-aware representation generalizes well beyond affinity regression, making FIRM-DTI a versatile tool for various drug–target interaction tasks.
6. While the study shows promising results, the authors acknowledge limitations such as reliance on pretrained experts and the absence of 3D structural information. Future work may explore joint training of molecular and protein embeddings and incorporate additional structural data to further enhance the model's capabilities.
7. The code for FIRM-DTI is available, allowing researchers to reproduce and build upon this work. This open approach fosters collaboration and accelerates progress in the field of computational drug discovery.
📜Paper: arxiv.org/abs/2509.20693
#DrugDiscovery #BindingAffinity #DeepLearning #ComputationalBiology #FIRM-DTI
6
27
1,745
26 Aug 2025
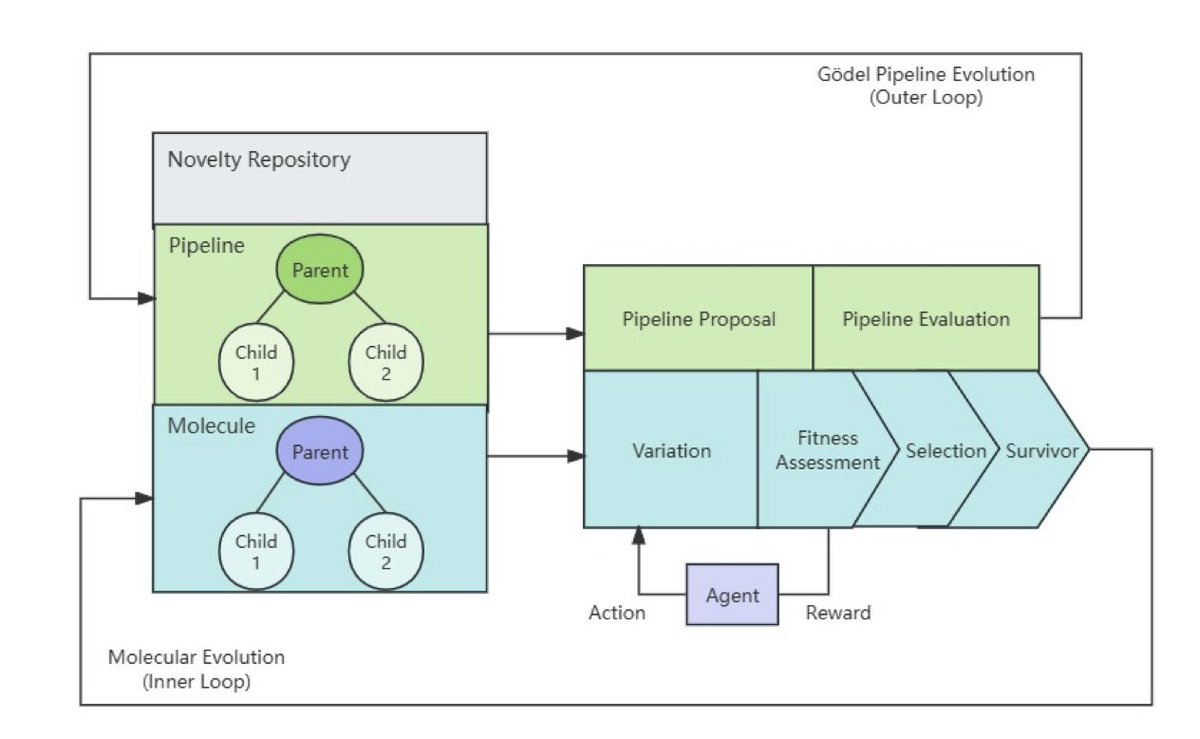
The Darwin–Gödel Drug Discovery Machine (DGDM): A Self-Improving AI Framework
1. The Darwin–Gödel Drug Discovery Machine (DGDM) is a novel AI framework designed to revolutionize drug discovery by integrating generative molecular design and evolution with adaptive meta-learning. This dual-loop architecture optimizes molecules through a Darwinian evolutionary process guided by reinforcement learning, while also adaptively modifying the discovery pipeline itself for continuous improvement.
2. Unlike traditional drug discovery pipelines that remain static, DGDM employs an inner loop for molecular optimization, treating it as a Darwinian evolutionary process. Candidate molecules generated by AI are evolved through search and feedback, with fitness assessment outcomes serving as reward signals for constraint-aware selection. This approach allows for broad exploration of chemical space while steering evolution toward higher-quality candidates.
3. The outer loop of DGDM is inspired by the Gödel machine, which proposes modifications to the discovery pipeline itself. Unlike the original Gödel machine that requires formal proofs of improvement, DGDM uses statistical validation to bound risk and ensure reliable progress. This data-driven approach ensures that only modifications meeting user-defined criteria are accepted, with a small, theoretically justified threshold for error.
4. DGDM is fully compatible with modern structural biology tools, including AlphaFold, and supports evaluation through docking, binding affinity prediction, and ADMET profiling. In a proof-of-concept study, DGDM improved the median binding affinity of candidate ligands from -4.457 to -5.422 kcal/mol while maintaining 100% drug-likeness and novelty. These results highlight the potential of bounded-risk, self-improving AI to accelerate drug discovery.
5. The framework is designed to be adaptable and scalable, with the potential to integrate experimental binding data and wet-lab feedback in future work. This could lead to a fully closed-loop discovery engine that combines computation with experimental validation, ultimately accelerating biomedical innovation with transparency and rigor.
6. The study acknowledges limitations, including the restricted evaluation to a small ligand panel and a single protein target. Future work should broaden benchmarking, incorporate rescoring and molecular dynamics, and integrate experimental binding data. Responsible deployment requires transparent benchmarking, reproducibility, and robust governance.
7. The code for DGDM is open-sourced, allowing researchers to explore and build upon this innovative framework. This open approach fosters collaboration and accelerates the development of new tools and techniques in AI-driven drug discovery.
💻Code: github.com/deep-geo/DGDM
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #AIFramework #SelfImprovingAI #AlphaFold #BindingAffinity #MolecularDesign #ComputationalBiology
2
2
5
1,682
4 Jul 2025
IntFold: A Controllable Foundation Model for General and Specialized Biomolecular Structure Prediction
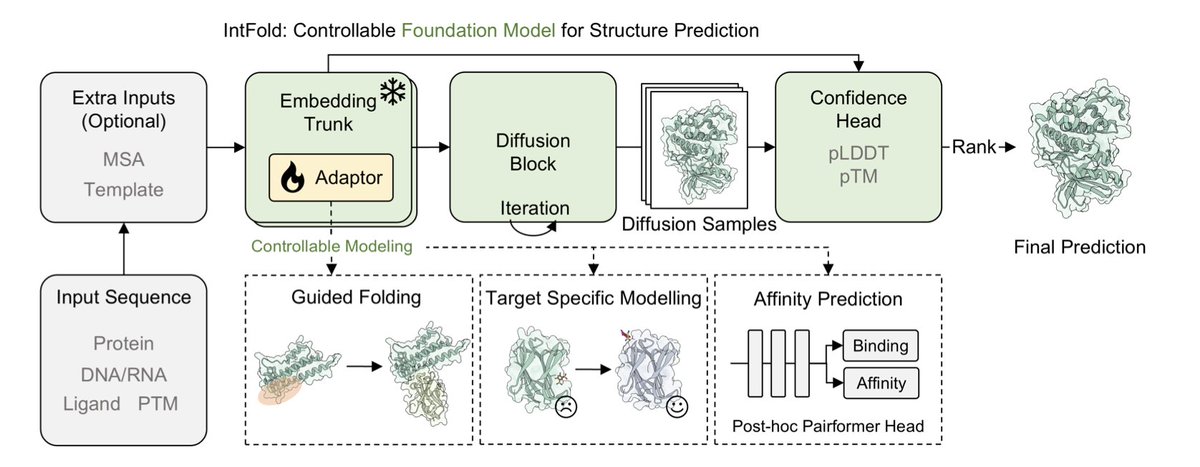
1.IntFold introduces a new class of biomolecular structure predictors—combining AlphaFold 3-level accuracy with fine-grained control over predictions. Its standout innovation: modular adapters enabling guided modeling of allosteric states, structural constraints, and binding affinities, critical for drug discovery.
2.On the FoldBench benchmark, IntFold matches AlphaFold 3 in protein monomer and protein-protein interaction prediction. It outperforms all other competitors—including Boltz-2 and HelixFold 3—across protein-ligand, antibody-antigen, and nucleic acid tasks.
3.A specialized variant, IntFold , further improves antibody-antigen docking (success rate 43.2%, nearly matching AlphaFold 3’s 47.9%) and protein-ligand interface predictions (61.8%), closing critical performance gaps in therapeutic contexts.
4.For CDK2, a classic allosteric kinase target, general models failed to capture inhibitor-induced conformations. IntFold’s fine-tuned adapter correctly identified 4 out of 5 rare allosteric states, without degrading accuracy on common structures—showcasing robust controllability.
5.By incorporating prior knowledge as structural constraints (e.g., known binding pockets or epitopes), IntFold drastically improves predictions. On antibody-antigen interfaces, success rates jumped from 37.6% to 69.0%—a major boost for immunological modeling.
6.IntFold delivers accurate binding affinity prediction using a downstream adapter. On DAVIS and BindingDB, it beats both structure-based and sequence-based baselines. On CASP16 targets, its predictions showed higher correlation with experimental affinities than Boltz-2.
7.The team developed a custom Triton-based attention kernel—FlashAttentionPairBias—more memory-efficient and faster than industry kernels from DeepSpeed and NVIDIA, enabling larger and more diverse training inputs.
8.A training-free, model-agnostic ranking method based on internal structural similarity improves prediction selection. For antibody-antigen targets, it raised success by 3% over random selection—offering a simple yet effective upgrade to multi-sample inference.
9.Training insights revealed sources of instability in large-scale biomolecular models. Solutions included layernorm tweaks, a “skip-and-recover” mechanism for exploding gradients, and carefully chosen initialization strategies, highlighting practical engineering know-how.
10.IntFold was trained on a rich and diverse dataset, including distilled predictions, curated affinity measurements, antibody-antigen distillations, and orthosteric/allosteric CDK2 complexes—laying a strong foundation for both generalization and specialization.
11.Despite its strengths, IntFold faces challenges typical of high-complexity models—such as cubic-time attention and imperfect performance on the hardest interface types. The team aims to improve scalability and expand applications into de novo protein design.
💻Code: github.com/IntelliGen-AI/Int…
📜Paper: arxiv.org/abs/2507.02025v1
#ProteinFolding #DrugDiscovery #AlphaFold #Biotech #ComputationalBiology #DeepLearning #IntFold #AntibodyDesign #MolecularModeling #BindingAffinity #AllostericPrediction
1
9
40
4,618
4 Jul 2025
IntFold: A Controllable Foundation Model for General and Specialized Biomolecular Structure Prediction
1.IntFold introduces a new class of biomolecular structure predictors—combining AlphaFold 3-level accuracy with fine-grained control over predictions. Its standout innovation: modular adapters enabling guided modeling of allosteric states, structural constraints, and binding affinities, critical for drug discovery.
2.On the FoldBench benchmark, IntFold matches AlphaFold 3 in protein monomer and protein-protein interaction prediction. It outperforms all other competitors—including Boltz-2 and HelixFold 3—across protein-ligand, antibody-antigen, and nucleic acid tasks.
3.A specialized variant, IntFold , further improves antibody-antigen docking (success rate 43.2%, nearly matching AlphaFold 3’s 47.9%) and protein-ligand interface predictions (61.8%), closing critical performance gaps in therapeutic contexts.
4.For CDK2, a classic allosteric kinase target, general models failed to capture inhibitor-induced conformations. IntFold’s fine-tuned adapter correctly identified 4 out of 5 rare allosteric states, without degrading accuracy on common structures—showcasing robust controllability.
5.By incorporating prior knowledge as structural constraints (e.g., known binding pockets or epitopes), IntFold drastically improves predictions. On antibody-antigen interfaces, success rates jumped from 37.6% to 69.0%—a major boost for immunological modeling.
6.IntFold delivers accurate binding affinity prediction using a downstream adapter. On DAVIS and BindingDB, it beats both structure-based and sequence-based baselines. On CASP16 targets, its predictions showed higher correlation with experimental affinities than Boltz-2.
7.The team developed a custom Triton-based attention kernel—FlashAttentionPairBias—more memory-efficient and faster than industry kernels from DeepSpeed and NVIDIA, enabling larger and more diverse training inputs.
8.A training-free, model-agnostic ranking method based on internal structural similarity improves prediction selection. For antibody-antigen targets, it raised success by 3% over random selection—offering a simple yet effective upgrade to multi-sample inference.
9.Training insights revealed sources of instability in large-scale biomolecular models. Solutions included layernorm tweaks, a “skip-and-recover” mechanism for exploding gradients, and carefully chosen initialization strategies, highlighting practical engineering know-how.
10.IntFold was trained on a rich and diverse dataset, including distilled predictions, curated affinity measurements, antibody-antigen distillations, and orthosteric/allosteric CDK2 complexes—laying a strong foundation for both generalization and specialization.
11.Despite its strengths, IntFold faces challenges typical of high-complexity models—such as cubic-time attention and imperfect performance on the hardest interface types. The team aims to improve scalability and expand applications into de novo protein design.
💻Code: github.com/IntelliGen-AI/Int…
📜Paper: arxiv.org/abs/2507.02025v1
#ProteinFolding #DrugDiscovery #AlphaFold #Biotech #ComputationalBiology #DeepLearning #IntFold #AntibodyDesign #MolecularModeling #BindingAffinity #AllostericPrediction

1
4
1,368