Don’t Retrain, Just Reuse: Recovering Dual-Target Molecules from Single-Target Diffusion Models
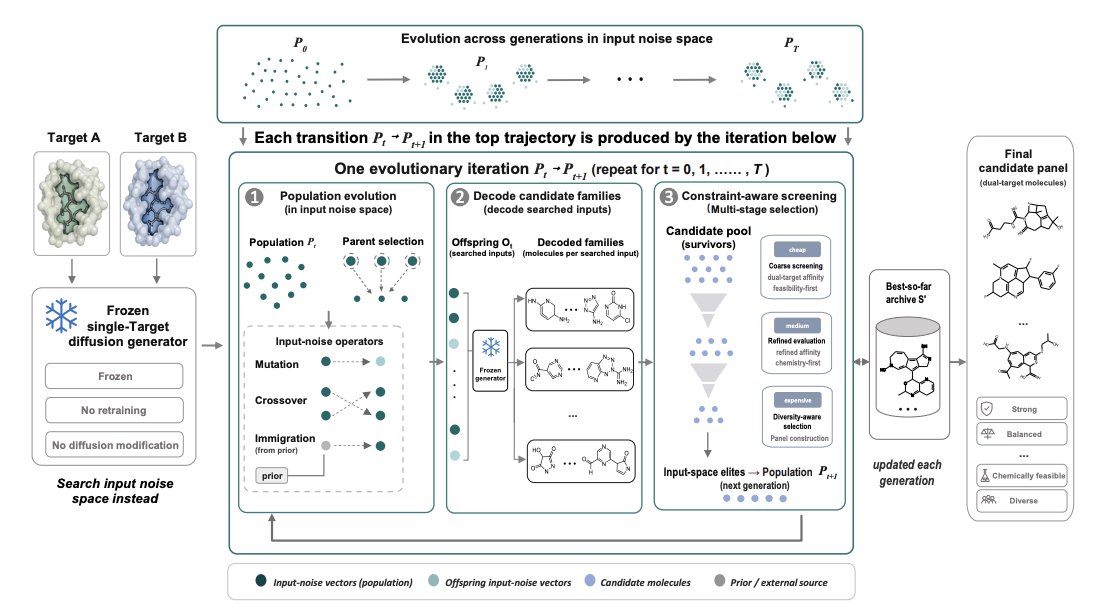
1. The paper asks a practical question in structure-based drug design: instead of retraining or adding sampling-time guidance for dual-target generation, can dual-target ligands be recovered by searching the input noise space of a frozen single-target diffusion model, keeping both parameters and denoising dynamics unchanged?
2. It formalizes dual-target design as constrained multi-objective optimization over searched noise inputs and a final candidate panel, jointly optimizing (i) dual-target affinity with explicit balance, (ii) chemical quality (drug-likeness and synthesizability), and (iii) set-level diversity, under hard feasibility constraints.
3. The proposed method, REUSE, is a hierarchical evolutionary search in the generator’s input noise space: maintain a population of noise vectors, generate offspring via mutation/crossover/immigration, decode each noise vector into a small “molecular family,” and use family-level evidence (not a single sample) to score and evolve the population toward regions that reliably yield dual-target candidates.
4. A key design choice is family-based fitness: each noise input is evaluated by aggregating scores of the top-ranked molecules within its decoded family, reducing sensitivity to stochastic decoding and favoring noise regions that consistently produce good candidates rather than one-off lucky samples.
5. REUSE uses cost-aware multi-stage environmental selection (coarse-to-fine): cheap docking/chemistry proxies first filter large pools with feasibility-first ranking, then expensive high-fidelity docking is applied only to a reduced frontier; final output is a diverse panel (not a single molecule), with explicit similarity constraints to avoid redundancy.
6. Balance is enforced directly in the affinity objective using a penalty on cross-target score disparity, discouraging “one-target-only” solutions; chemistry control is incorporated via QED/SA-based terms plus hard floors, so optimization does not collapse into unrealistic high-docking-score artifacts.
7. On the Zhou et al. dual-target benchmark (12,917 target pairs, 438 targets), using TargetDiff as the frozen backbone, REUSE achieves the best reported docking-centered dual-target metrics: best average P-2 Vina Dock (-9.26), best Max Vina Dock (-8.64), and highest Dual High Affinity rate (58.3%), improving Dual High Affinity by 20.9 percentage points over the strongest prior baseline (MDRL).
8. Ablations isolate failure modes: removing input-space search sharply degrades dual-target recovery (Dual High Affinity 58.3% → 31.8%); removing balance hurts cross-target consistency; removing chemistry control can slightly improve docking but substantially worsens QED/SA, highlighting why multi-objective constraints matter.
9. The paper provides evidence that the frozen input space has exploitable local structure: high-quality noise “anchors” tend to have enriched high-quality neighbors, supporting why evolutionary local exploration (mutation) can work better than naive random sampling.
📜Paper: arxiv.org/abs/2605.25681
#ComputationalBiology #MachineLearning #DiffusionModels #DrugDiscovery #Polypharmacology #StructureBasedDrugDesign #MolecularGeneration #MultiObjectiveOptimization #EvolutionaryAlgorithms
8
18
1,649
SMDD-Bench: Can LLMs Solve Real-World Small Molecule Drug Design Tasks?
1. The paper introduces SMDD-Bench, a multi-turn, long-horizon benchmark designed to test whether LLM agents can handle realistic small-molecule drug design workflows, rather than single-turn chemistry QA. It contains 502 guaranteed-solvable task instances spanning diverse chemistries and 102 protein targets.
2. The most striking result: even the best tested model (GPT-5.4) solves only 40.2% overall, despite using a tool-enabled agent scaffold. Performance concentrates heavily in Lead Optimization, while 3D-heavy tasks (Interaction Point Discovery, Scaffold Hopping, Fragment Assembly) remain near-zero for most models.
3. SMDD-Bench covers five task types that map to core medicinal chemistry loops: 2D Pharmacophore Identification, Interaction Point Discovery (3D pocket hotspots), Scaffold Hopping (new scaffold, same binding mode), Lead Optimization (multi-property optimization under constraints), and Fragment Assembly (link/expand fragments while preserving poses).
4. A key methodological contribution is witness-aware task generation: for task types where solvability is not guaranteed by default (Scaffold Hopping, Lead Optimization, Fragment Assembly), each instance is constructed alongside a hidden “witness” molecule that is known to pass the same evaluation pipeline. This makes every task instance solvable by construction, enabling scale without manual curation.
5. Evaluation is fully automated and tool-grounded: RDKit and OpenBabel for chemistry checks, PLIP for interaction fingerprints, Boltz2 for co-folded protein–ligand structures plus binding probability/affinity, and ADMET-AI for 8 ADMET endpoints (e.g., hERG, CYP3A4, solubility, clearance, BBB, Ames). Agents operate under limited oracle budgets to mimic scarce wet-lab feedback.
6. Lead Optimization is formulated as constrained multi-objective optimization: improve up to 5 properties, hold up to 4 within tolerance bands, satisfy hard drug-likeness constraints (MW, logP, TPSA, HBD/HBA, rotatable bonds, charge, SA score, PAINS/Brenk/NIH alerts), and remain in-series via a Tanimoto similarity constraint to the reference (≥0.7).
7. The benchmark also surfaces a practical agent failure mode: enumeration vs. selection. By retrospectively scoring all SMILES an agent mentioned (including those it did not submit to oracles), the authors show “recovered” success rates can be much higher—especially for Scaffold Hopping—implying agents often generate viable candidates but fail to choose them without extra oracle calls.
8. Output novelty is quantified against major public chemistry databases (ChEMBL, SureChEMBL, PubChem, BindingDB). Many models’ successful submissions are largely novel by this conservative check, but novelty alone is not sufficient: agents still struggle to satisfy 3D interaction constraints and multi-turn planning under budget.
9. Diversity is evaluated via SMDD-Bench Diversity (20 hard Lead Optimization tasks, 10 runs each). Models frequently converge to the same solutions; the benchmark reports unique-success counts and pairwise similarity, highlighting that “many parallel agents” may not explore chemical space as broadly as desired in real campaigns.
10. The authors provide SMDD-Bench Lite (100 instances) to reduce compute barriers, and document common failure modes beyond raw accuracy: lack of cross-turn SAR synthesis (re-testing disqualified motifs), incoherent multi-turn planning (not building on previous partial successes), and tool-specific code errors (e.g., malformed Boltz calls, RDKit conversion issues).
📜Paper: arxiv.org/abs/2605.21740
#LLM #DrugDiscovery #MedicinalChemistry #Cheminformatics #Benchmark #Agents #ADMET #StructureBasedDrugDesign #FragmentBasedDrugDiscovery #ComputationalBiology

4
14
1,468

May 12
Attention GPCR, Kinases and membrane protein enthusiast. Please check this 💊⚛️
Assessing State-Specific Accuracy of Cofolding Models for Kinases and GPCRs
biorxiv.org/content/10.64898…
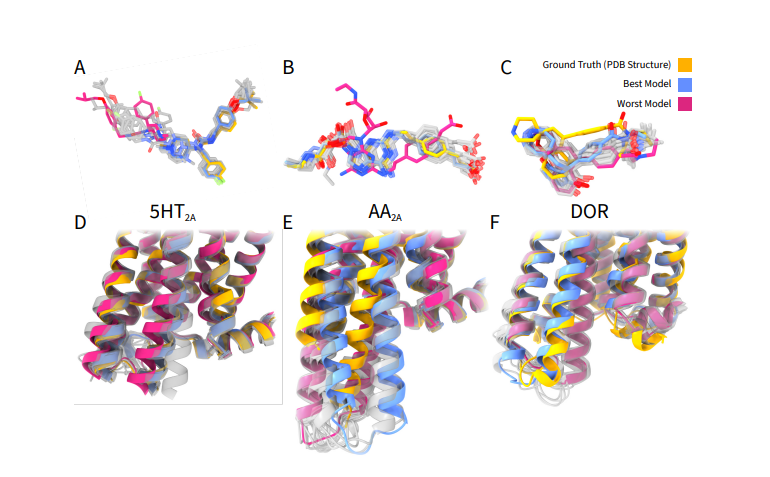
This study systematically evaluates the ability of modern AI-driven cofolding models to predict not only protein–ligand binding poses, but also the correct functional conformational states of dynamic drug targets. Benchmarking four leading models—#AlphaFold3, #RosettaFold3, #Boltz-2, and #Chai1—across kinases and Class A GPCRs, the authors reveal a major limitation termed “conformational decoupling,” where ligand placement can appear highly accurate while the broader protein activation state is incorrectly modeled. Although cofolding methods effectively remodel local binding pockets and often achieve low ligand RMSD for orthosteric binders, they frequently fail to recover critical structural hallmarks such as kinase activation-loop geometries or GPCR intracellular conformations that define signaling state and pharmacological behavior.
The study further shows that introducing state-biased templates and filtered MSAs can improve conformational recovery in select cases, but these strategies remain inconsistent and target-dependent. Importantly, all models exhibited a strong weakness in predicting cryptic or allosteric binding sites, with allosteric ligands repeatedly misplaced into canonical orthosteric pockets despite accurate global protein folds. Confidence metrics such as pLDDT were also poor indicators of functional-state accuracy, emphasizing that apparent structural confidence does not guarantee biologically meaningful conformations. Overall, the work highlights that while current cofolding approaches are powerful for orthosteric pose prediction and induced-fit-like pocket remodeling, they remain unreliable for state-selective modeling and allosteric drug discovery—an important challenge for the future of AI-driven structure-based drug design.
💊 #DrugDiscovery 🧠 #BioAI 🔬 #StructuralBiology 🧪 #ProteinFolding 🧫 #GPCR 🎯 #Kinase 🧩 #ProteinLigand 💡 #AllostericModulation 📊 #MolecularModeling 🧱 #StructureBasedDrugDesign 💉 #Pharmacology 🌐 #SystemsBiology 🔍 #MedicinalChemistry
3
8
40
2,188
AgenticPosesRanker: An agentic AI framework for physically grounded ranking of protein–ligand docking poses
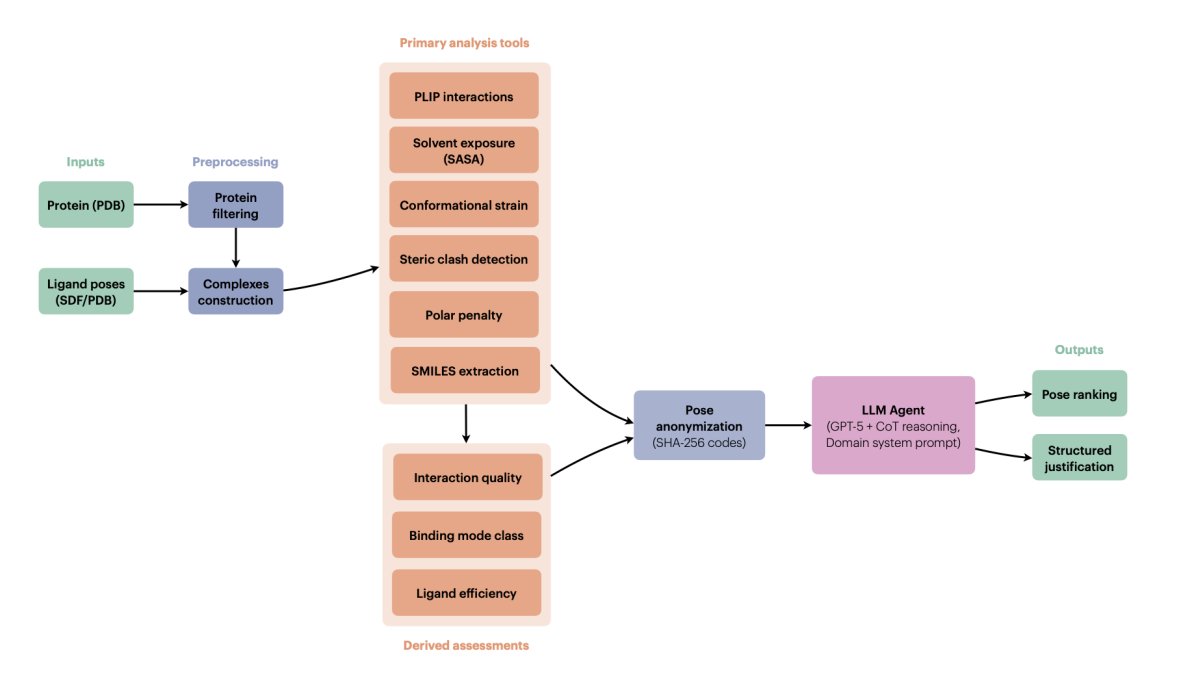
1. The paper proposes a tool-augmented LLM “pose judge” that ranks docking poses by explicit multi-criteria reasoning over physically interpretable metrics, rather than learning or fitting a new scoring function.
2. AgenticPosesRanker combines 6 deterministic analysis tools (PLIP interaction fingerprinting, SASA/burial, MMFF94-based conformational strain, steric clash detection, unsatisfied buried polar atom penalty, and SMILES identity checks) plus 3 derived assessments (interaction quality scoring, binding-mode classification, ligand efficiency).
3. Key design choice: keep each physicochemical observable separate and interpretable, then let the LLM integrate conflicting evidence (e.g., strong interactions vs. high strain, deep burial vs. polar desolvation risk) with a structured decision rubric instead of fixed weights.
4. The agent uses GPT-5 with chain-of-thought enabled and a long domain-specific system prompt that encodes tool documentation, thresholds, and a hierarchical ranking policy (e.g., binding mode and interaction quality prioritized; strain/clashes/polar penalties as discriminators; “surface binders should never rank first”).
5. To reduce name-based leakage (e.g., pose_01 implying docking-score order), poses are anonymized via deterministic SHA-256-derived 8-character base-36 codes; tool outputs are sanitized to remove original identifiers before being passed to the LLM, then de-anonymized after inference.
6. Benchmark: 10 protein–ligand systems, 162 poses, curated to be balanced “by construction” between cases where Smina ranks a near-native pose correctly and cases where Smina fails. Ground truth is RMSD to crystallographic pose.
7. Main result: 50.0% best-pose accuracy on the 10-system benchmark, matching the design-fixed Smina baseline (50.0%) and far above a uniformly random baseline (7.7%; p < 0.001, one-sided exact binomial test). The aggregate hides symmetry: 80% retention of Smina successes (4/5) but only 20% recovery of Smina failures (1/5).
8. A notable methodological contribution is “decision attribution” analysis: the paper compares the agent’s self-reported tool weighting against objective metric separations for the selected pose, finding strong alignment (median Spearman ρ = 0.83) across both correct and incorrect outcomes—suggesting the limiting factor is tool-suite coverage, not inconsistent reasoning.
9. The work emphasizes interpretability and auditability: instead of an opaque scalar score, the output includes metric-by-metric justification and structured comparative analysis, positioning the agent as an interpretable curation layer for late-stage pose refinement rather than a guaranteed accuracy upgrade over classical scoring.
📜Paper: arxiv.org/abs/2605.03707
#computationalbiology #computationalchemistry #moleculardocking #structurebaseddrugdesign #LLM #agenticAI #interpretableAI #drugdiscovery #proteinligand #cheminformatics
2
1,045
AgenticPosesRanker: An agentic AI framework for physically grounded ranking of protein–ligand docking poses
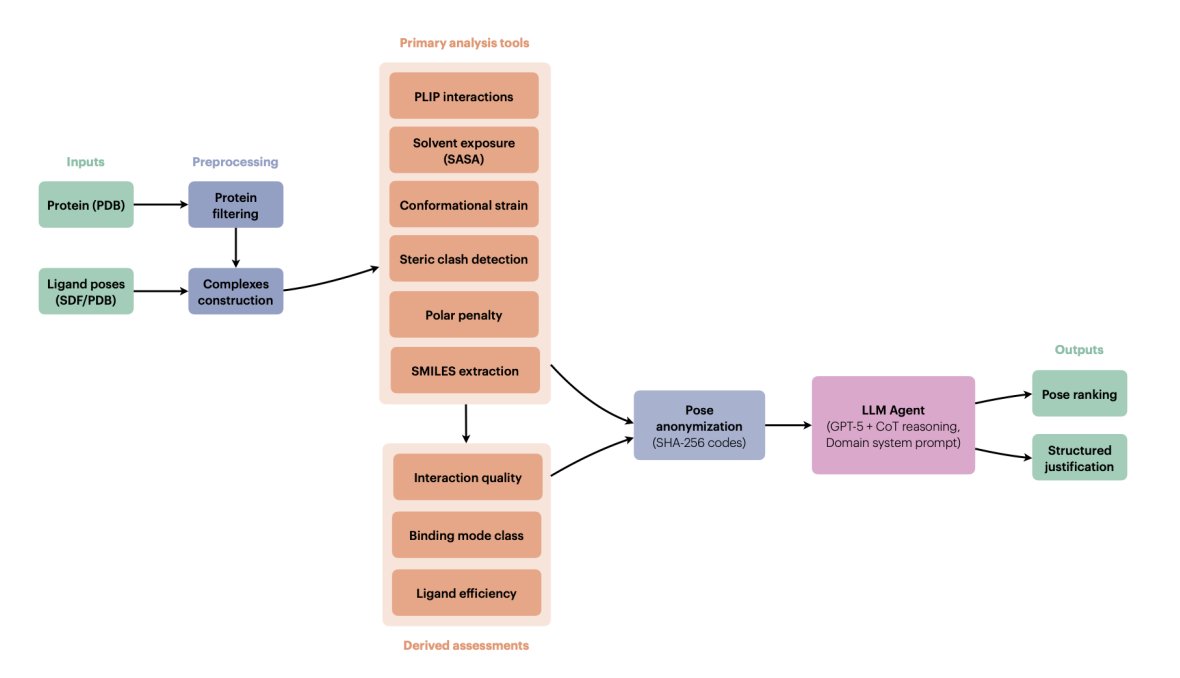
1. The paper proposes a tool-augmented LLM “pose judge” that ranks docking poses by explicit multi-criteria reasoning over physically interpretable metrics, rather than learning or fitting a new scoring function.
2. AgenticPosesRanker combines 6 deterministic analysis tools (PLIP interaction fingerprinting, SASA/burial, MMFF94-based conformational strain, steric clash detection, unsatisfied buried polar atom penalty, and SMILES identity checks) plus 3 derived assessments (interaction quality scoring, binding-mode classification, ligand efficiency).
3. Key design choice: keep each physicochemical observable separate and interpretable, then let the LLM integrate conflicting evidence (e.g., strong interactions vs. high strain, deep burial vs. polar desolvation risk) with a structured decision rubric instead of fixed weights.
4. The agent uses GPT-5 with chain-of-thought enabled and a long domain-specific system prompt that encodes tool documentation, thresholds, and a hierarchical ranking policy (e.g., binding mode and interaction quality prioritized; strain/clashes/polar penalties as discriminators; “surface binders should never rank first”).
5. To reduce name-based leakage (e.g., pose_01 implying docking-score order), poses are anonymized via deterministic SHA-256-derived 8-character base-36 codes; tool outputs are sanitized to remove original identifiers before being passed to the LLM, then de-anonymized after inference.
6. Benchmark: 10 protein–ligand systems, 162 poses, curated to be balanced “by construction” between cases where Smina ranks a near-native pose correctly and cases where Smina fails. Ground truth is RMSD to crystallographic pose.
7. Main result: 50.0% best-pose accuracy on the 10-system benchmark, matching the design-fixed Smina baseline (50.0%) and far above a uniformly random baseline (7.7%; p < 0.001, one-sided exact binomial test). The aggregate hides symmetry: 80% retention of Smina successes (4/5) but only 20% recovery of Smina failures (1/5).
8. A notable methodological contribution is “decision attribution” analysis: the paper compares the agent’s self-reported tool weighting against objective metric separations for the selected pose, finding strong alignment (median Spearman ρ = 0.83) across both correct and incorrect outcomes—suggesting the limiting factor is tool-suite coverage, not inconsistent reasoning.
9. The work emphasizes interpretability and auditability: instead of an opaque scalar score, the output includes metric-by-metric justification and structured comparative analysis, positioning the agent as an interpretable curation layer for late-stage pose refinement rather than a guaranteed accuracy upgrade over classical scoring.
📜Paper: arxiv.org/abs/2605.03707
#computationalbiology #computationalchemistry #moleculardocking #structurebaseddrugdesign #LLM #agenticAI #interpretableAI #drugdiscovery #proteinligand #cheminformatics
2
3
863
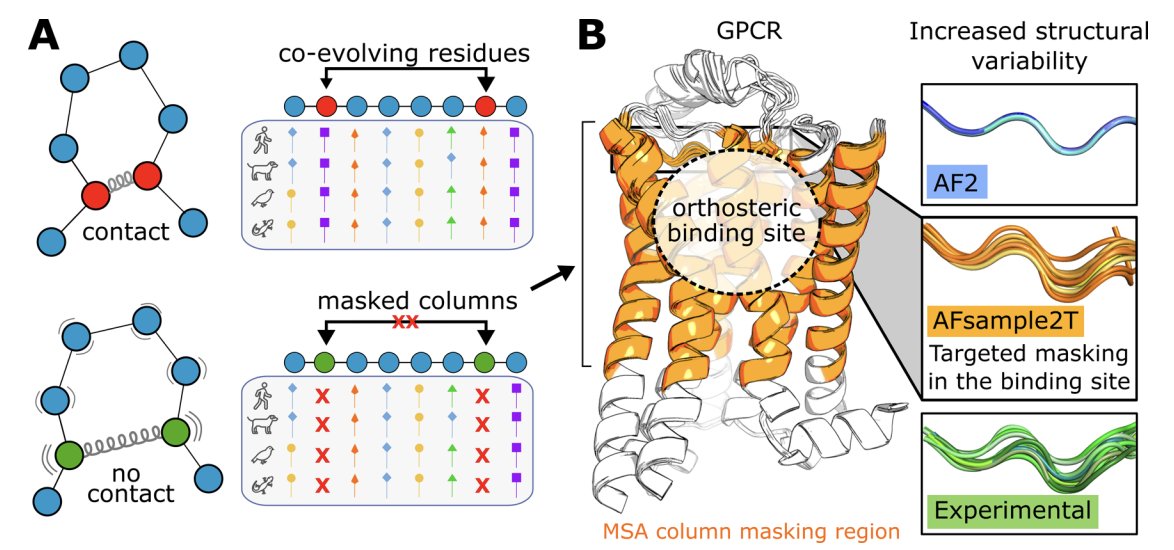
Improving AlphaFold2 Performance in Virtual Screens Targeting GPCRs by Enhancing Binding-Site Conformational Sampling
1. The paper introduces AFsample2T, a targeted AlphaFold2 sampling strategy that boosts virtual screening performance for GPCRs by generating diverse binding-site conformations rather than relying on a single “best” structure.
2. Core idea: mask selected MSA columns only in the orthosteric-site region (extracellular-facing TM segments part of EL2) to weaken local coevolution constraints and encourage alternative pocket geometries, while keeping the rest of the receptor well-constrained.
3. AFsample2T contrasts with global masking (AFsample2), which reduced binding-site accuracy for GPCRs here (AUC 0.38). Targeted masking preserved fold quality while improving pocket modeling, showing that “where to perturb” matters as much as “how much to perturb”.
4. The authors benchmarked 10 class A GPCRs using 61 curated experimental binding-site structures (from an initial pool of 119 PDB structures) and generated 1,000 models per receptor to quantify how often predicted pockets match experimental ones within 1–2 Å side-chain RMSD (symmetry-aware).
5. Moderate targeted masking improved binding-site accuracy: default AF2 AUC 0.54; adding dropout without masking AUC 0.57; targeted masking at 10–30% reached AUC ~0.59–0.61. Too much masking (50%) degraded secondary structure and collapsed performance (AUC 0.43).
6. The best-performing ensemble (AFsample2T) mixes 250 models each from 0%, 10%, 20%, and 30% masking (with dropout), yielding AUC 0.63 and capturing 73.8% of experimental binding sites at 1.5 Å RMSD vs 60.7% for AF2 (a 22% relative gain at that threshold).
7. A key mechanistic improvement is realistic binding-site plasticity. Median binding-site side-chain RMSF increased from 0.15 Å (AF2; overly rigid pockets) to 0.45 Å (AFsample2T), approaching experimental variability (median 0.58 Å). Backbone RMSF similarly moved from 0.10 Å (AF2) to 0.28 Å (AFsample2T), close to experiment (0.30 Å).
8. AFsample2T also mitigates a known docking issue: AF2 often predicts narrow/collapsed pockets. Across receptors, mean pocket volume increased (209 → 218 Å3) and the “top 1% most open” pockets expanded substantially (272 → 389 Å3), closer to experimental pockets (mean 256 Å3). This was especially relevant for μ-opioid receptor, where AF2 pockets were too collapsed.
9. Virtual screening evaluation used DOCK3.8 with ChEMBL actives (52–202 per receptor) and property-matched ZINC20 decoys, totaling extremely large-scale docking (reported as >240 trillion complexes scored). Rigid-receptor docking was used, making pocket microstates critical.
10. Ensemble screening ligand-guided model selection is the practical win: while median enrichment of AF2-based models remained below experimental structures, the top 1% AFsample2T models improved early enrichment (mean aLogAUC top 1%: 10.8 → 12.9; mean EF1% top 1%: 7.5 → 9.6). In some targets (e.g., TAAR1, μ-opioid receptor), best AFsample2T models approached top experimental-structure performance.
11. The paper provides a workflow for prospective use: generate ≥250 AFsample2T models for the relevant receptor state, dock a ligand/decoy control set to compute enrichment, select ~top 1% models, manually inspect key interactions/poses, then proceed to large-library prospective screening.
12. Modeling receptor state is handled explicitly: inactive models use receptor sequence alone; active models are generated by cofolding receptor with heterotrimeric G protein sequences via AF2-Multimer, capturing hallmark TM6 movements and separating “state sampling” from “pocket microstate sampling”.
💻Code: github.com/wallnerlab/AFsamp…
📜Paper: doi.org/10.1021/acs.jcim.6c0…
#AlphaFold2 #GPCR #VirtualScreening #Docking #StructureBasedDrugDesign #ComputationalChemistry #Bioinformatics #ProteinStructure #DrugDiscovery #MachineLearning
15
90
4,981

Apr 25
@SuperWarmai just published my #article📝 on why @MedSynthTM is the #future🔮 of #DrugDesign💊: it’s #3D🧬
medium.com/strtupboost/the-f…
#AI🤖
#StructureBasedDrugDesign💊
#SBDD💊
#DrugDiscovery🔬
#AppleVisionPro🥽
#SpatialComputing🥽
#QuantumComputing⚛️
@Apple
@spatialreport

2
4
190
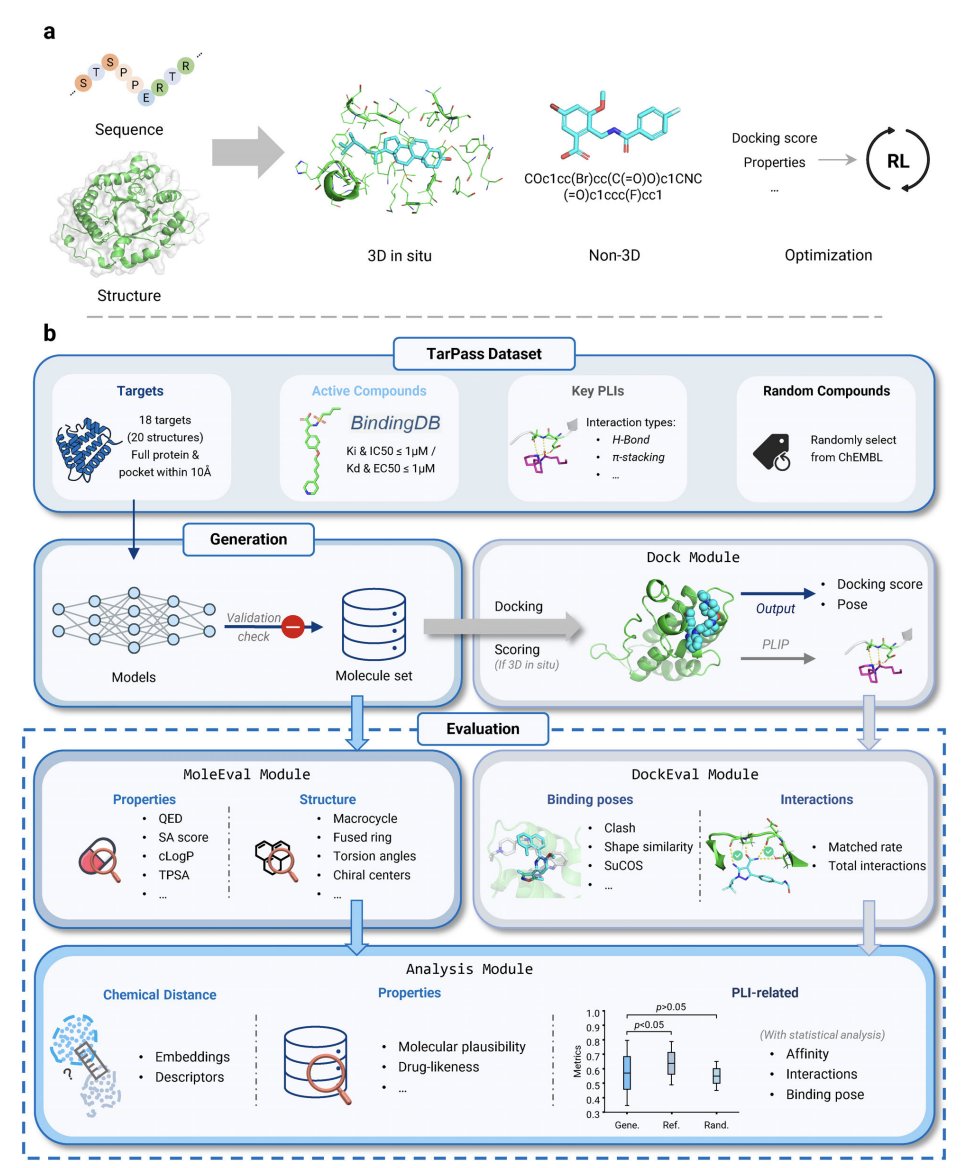
Revisiting Target-Aware de novo Molecular Generation with TarPass: Between Rational Design and Texas Sharpshooter
1. The paper argues that many “target-aware” de novo generators may not truly use target information, but instead risk a Texas Sharpshooter pattern: retrospectively rationalizing outputs using coarse metrics (e.g., docking) and cherry-picked examples.
2. To address this, the authors introduce TarPass, a curated benchmark designed for fair, target-grounded evaluation across paradigms. It includes 18 well-studied, pharmaceutically relevant targets (20 structures total), expert-annotated key interactions, and ~1000 experimentally validated actives per target (from BindingDB), plus a ChEMBL-random baseline to test whether models beat “just sample from a drug-like database.”
3. TarPass is explicitly built to reduce data leakage: targets are time-split (post-2019) and selected to avoid overlap with common structure–ligand training sets (CrossDocked2020, PDBbind). The benchmark frames generalization realistically as “within druggable families” (e.g., kinases) rather than assuming entirely novel folds.
4. The evaluation is holistic and standardized: generate up to 1000 unique molecules/target, run a consistent docking workflow (with special handling for 3D in situ initial poses), then score both protein–ligand interactions (PLIs) and molecular plausibility (validity, drug-likeness, synthesizability, structural alerts, and chemical-distance behavior).
5. 15 representative methods are benchmarked across three paradigms: non-3D (DeepBlock, DRAGONFLY, SimpleSBDD, TamGen), 3D in situ (DiffSBDD, DrugFlow, IPDiff, Lingo3DMol, MolCraft, PocketFlow, SurfGen, TargetDiff), and optimization-based variants (DrugFlow-PA, MolPilot, REINVENT). The study also reports practical deployability: runtime, validity, uniqueness, and input-structure compatibility.
6. Key PLI finding: 3D in situ methods show only a modest average advantage in docking/interaction metrics, and many do not significantly outperform the ChEMBL-random baseline across targets. Only a small subset of methods shows consistent gains, and even then performance can be sensitive to conditions like reliance on an input ligand (raising concerns about robustness/generalization).
7. Interaction recovery is used as a stricter test than docking score alone. Even reference ligands achieve only ~51% exact match (limited by docking/PLIP constraints), but most models perform near random on exact match and match ratio; only a few (notably including DrugFlow/MolCraft and optimized variants) approach reference-like interaction recovery.
8. Pose realism remains a bottleneck for 3D in situ generation: initial conformations frequently contain steric clashes, centroid placement errors correlate strongly with reduced interaction recovery, and certain targets expose systematic failure modes (e.g., incomplete pocket definitions causing clashes; metal coordination such as Zn in HDAC6 being mishandled or unsupported by some models).
9. Plausibility/drug-likeness trade-off: non-3D models (often benefiting from broader pretraining) tend to generate more drug-like and synthesizable molecules (higher QED, better SA scores, fewer medicinal-chemistry alerts) but show weaker target specificity in PLIs. Many graph-based 3D in situ models overproduce implausible stereochemistry and overly complex ring systems (e.g., highly fused rings), harming synthetic feasibility.
10. The paper proposes a practical post-processing strategy: a multi-tier virtual screening workflow that applies hard filters across PLIs plausibility drug-likeness, followed by softer refinement (experience-based filters, optional clustering/MD). In case studies (JAK2/TYK2), hard filters reduce libraries to ~10% and later steps downscale to ~20–30 candidates, yielding some enrichment—but still highlighting that filtering cannot substitute for improving pose accuracy, interaction fidelity, and plausibility in the generators themselves.
📜Paper: doi.org/10.1002/advs.75411
#ComputationalBiology #DrugDiscovery #GenerativeAI #MolecularGeneration #StructureBasedDrugDesign #Benchmarking #Docking #Cheminformatics #MachineLearning

2
1,042
Revisiting Target-Aware de novo Molecular Generation with TarPass: Between Rational Design and Texas Sharpshooter
1. The paper argues that many “target-aware” de novo generators may not truly use target information, but instead risk a Texas Sharpshooter pattern: retrospectively rationalizing outputs using coarse metrics (e.g., docking) and cherry-picked examples.
2. To address this, the authors introduce TarPass, a curated benchmark designed for fair, target-grounded evaluation across paradigms. It includes 18 well-studied, pharmaceutically relevant targets (20 structures total), expert-annotated key interactions, and ~1000 experimentally validated actives per target (from BindingDB), plus a ChEMBL-random baseline to test whether models beat “just sample from a drug-like database.”
3. TarPass is explicitly built to reduce data leakage: targets are time-split (post-2019) and selected to avoid overlap with common structure–ligand training sets (CrossDocked2020, PDBbind). The benchmark frames generalization realistically as “within druggable families” (e.g., kinases) rather than assuming entirely novel folds.
4. The evaluation is holistic and standardized: generate up to 1000 unique molecules/target, run a consistent docking workflow (with special handling for 3D in situ initial poses), then score both protein–ligand interactions (PLIs) and molecular plausibility (validity, drug-likeness, synthesizability, structural alerts, and chemical-distance behavior).
5. 15 representative methods are benchmarked across three paradigms: non-3D (DeepBlock, DRAGONFLY, SimpleSBDD, TamGen), 3D in situ (DiffSBDD, DrugFlow, IPDiff, Lingo3DMol, MolCraft, PocketFlow, SurfGen, TargetDiff), and optimization-based variants (DrugFlow-PA, MolPilot, REINVENT). The study also reports practical deployability: runtime, validity, uniqueness, and input-structure compatibility.
6. Key PLI finding: 3D in situ methods show only a modest average advantage in docking/interaction metrics, and many do not significantly outperform the ChEMBL-random baseline across targets. Only a small subset of methods shows consistent gains, and even then performance can be sensitive to conditions like reliance on an input ligand (raising concerns about robustness/generalization).
7. Interaction recovery is used as a stricter test than docking score alone. Even reference ligands achieve only ~51% exact match (limited by docking/PLIP constraints), but most models perform near random on exact match and match ratio; only a few (notably including DrugFlow/MolCraft and optimized variants) approach reference-like interaction recovery.
8. Pose realism remains a bottleneck for 3D in situ generation: initial conformations frequently contain steric clashes, centroid placement errors correlate strongly with reduced interaction recovery, and certain targets expose systematic failure modes (e.g., incomplete pocket definitions causing clashes; metal coordination such as Zn in HDAC6 being mishandled or unsupported by some models).
9. Plausibility/drug-likeness trade-off: non-3D models (often benefiting from broader pretraining) tend to generate more drug-like and synthesizable molecules (higher QED, better SA scores, fewer medicinal-chemistry alerts) but show weaker target specificity in PLIs. Many graph-based 3D in situ models overproduce implausible stereochemistry and overly complex ring systems (e.g., highly fused rings), harming synthetic feasibility.
10. The paper proposes a practical post-processing strategy: a multi-tier virtual screening workflow that applies hard filters across PLIs plausibility drug-likeness, followed by softer refinement (experience-based filters, optional clustering/MD). In case studies (JAK2/TYK2), hard filters reduce libraries to ~10% and later steps downscale to ~20–30 candidates, yielding some enrichment—but still highlighting that filtering cannot substitute for improving pose accuracy, interaction fidelity, and plausibility in the generators themselves.
📜Paper: doi.org/10.1002/advs.75411
#ComputationalBiology #DrugDiscovery #GenerativeAI #MolecularGeneration #StructureBasedDrugDesign #Benchmarking #Docking #Cheminformatics #MachineLearning
3
13
1,017
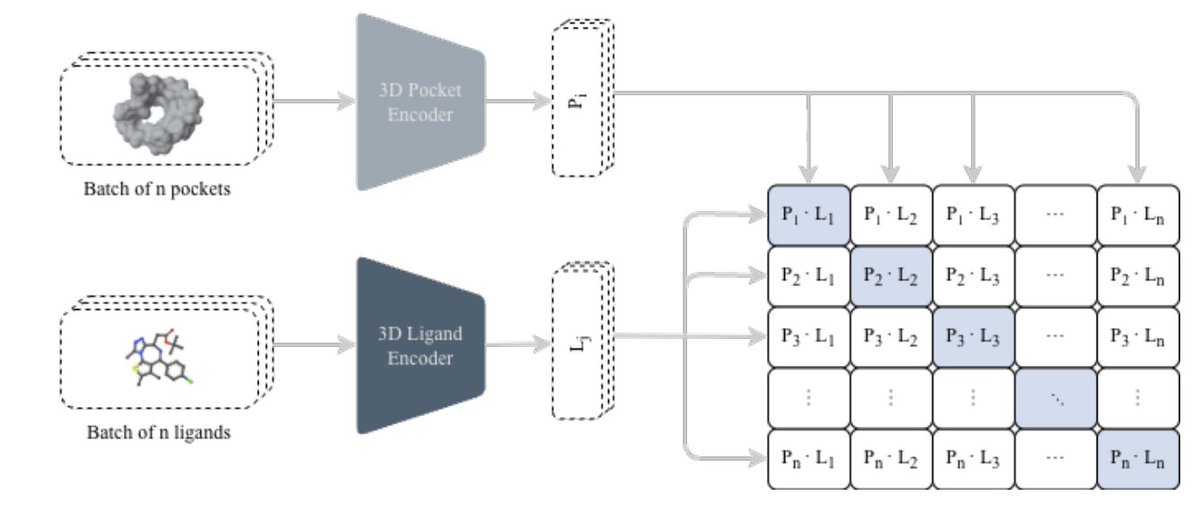
Structure-guided molecular design with contrastive 3D protein–ligand learning
1 The paper proposes a unified pipeline that connects two usually separate steps in structure-based drug discovery: (i) fast 3D protein–ligand compatibility retrieval via contrastive learning, and (ii) de novo molecule generation via an autoregressive chemical language model, while explicitly steering outputs toward purchasable/synthetically accessible commercial spaces.
2 At the core is CLIPP-SET: an SE(3)-equivariant Transformer encoder that maps 3D pockets and 3D ligands into a shared embedding space, enabling cosine-similarity screening without docking ultra-large libraries and without target-specific fine-tuning (zero-shot).
3 A key technical detail is how they handle “false negatives” in contrastive learning caused by pocket collisions (many ligands bind the same pocket): they use Collision-Free InfoNCE (CF-InfoNCE), selecting (for pocket→ligand) the strongest-affinity ligand among those sharing the same pocket as the positive, rather than treating all off-diagonal pairs as negatives.
4 On LIT-PCBA zero-shot virtual screening (15 targets), CLIPP-SET pocket-based screening shows particularly strong early enrichment: best BEDROC, EF(0.5%), and EF(1%) among compared baselines, indicating it prioritizes actives near the top of the ranked list even if broader AUROC/EF(5%) can favor some docking-based methods.
5 The work then tests “realistic scale” retrieval on Enamine REAL (5.9B compounds): pocket-only queries (no reference ligand) retrieve top-100 candidates per target with higher predicted pIC50 on 13/15 targets and much higher internal diversity than 2D Morgan fingerprint search, suggesting the embedding captures binding-relevant geometry rather than just 2D similarity.
6 Because ligands and pockets share the same embedding space, the same system supports ligand-based search for scaffold hopping: given a known active, it retrieves molecules with high 3D shape/pharmacophore similarity but low 2D similarity (lower Tanimoto than Morgan FP), aligning with the goal of finding chemically novel scaffolds that preserve 3D binding features.
7 For generation, they introduce an MCLM (multimodal chemical language model): a Llama2-style autoregressive SMILES decoder conditioned by prepending a learned structural embedding token (from the frozen contrastive encoder) plus a learned dataset token [ds] that controls which chemical space distribution to emulate.
8 The dataset token is the mechanism for “commercial space steering”: trained on a 287M conformer–SMILES corpus labeled by source (PubChem, Enamine Diversity, Mcule, ChEMBL, GEOM-drugs), the model can be prompted with an “Enamine” token at inference to bias outputs toward Enamine-like chemistry; an ablation shows the token increases nearest-neighbor similarity to Enamine REAL and raises the fraction of exact catalog matches.
9 In structure-conditioned de novo design on LIT-PCBA (100 molecules/target), ligand-conditioned generation achieves the highest predicted affinity metrics (affinity probability and predicted pIC50) among evaluated methods, while pocket-conditioned generation (despite the decoder being trained only with ligand embeddings) remains competitive without reference ligands and yields the highest diversity, highlighting practical utility when only target structure is available.
📜Paper: arxiv.org/abs/2604.19562
#ComputationalBiology #Cheminformatics #DrugDiscovery #StructureBasedDrugDesign #GenerativeAI #MachineLearning #ProteinLigand #EquivariantNetworks #ContrastiveLearning #VirtualScreening
1
6
25
7,268
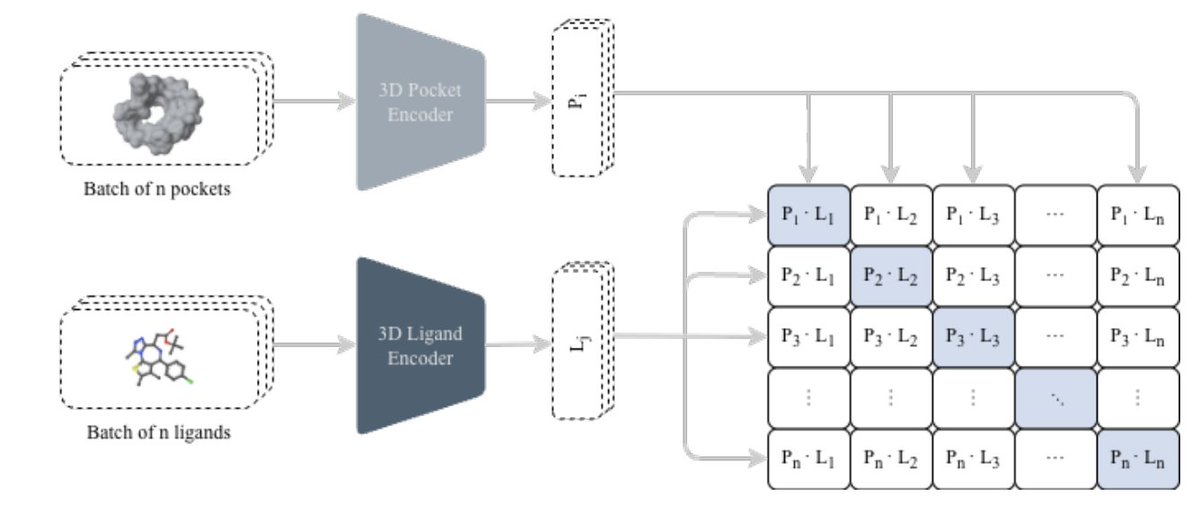
Structure-guided molecular design with contrastive 3D protein–ligand learning
1 The paper proposes a unified pipeline that connects two usually separate steps in structure-based drug discovery: (i) fast 3D protein–ligand compatibility retrieval via contrastive learning, and (ii) de novo molecule generation via an autoregressive chemical language model, while explicitly steering outputs toward purchasable/synthetically accessible commercial spaces.
2 At the core is CLIPP-SET: an SE(3)-equivariant Transformer encoder that maps 3D pockets and 3D ligands into a shared embedding space, enabling cosine-similarity screening without docking ultra-large libraries and without target-specific fine-tuning (zero-shot).
3 A key technical detail is how they handle “false negatives” in contrastive learning caused by pocket collisions (many ligands bind the same pocket): they use Collision-Free InfoNCE (CF-InfoNCE), selecting (for pocket→ligand) the strongest-affinity ligand among those sharing the same pocket as the positive, rather than treating all off-diagonal pairs as negatives.
4 On LIT-PCBA zero-shot virtual screening (15 targets), CLIPP-SET pocket-based screening shows particularly strong early enrichment: best BEDROC, EF(0.5%), and EF(1%) among compared baselines, indicating it prioritizes actives near the top of the ranked list even if broader AUROC/EF(5%) can favor some docking-based methods.
5 The work then tests “realistic scale” retrieval on Enamine REAL (5.9B compounds): pocket-only queries (no reference ligand) retrieve top-100 candidates per target with higher predicted pIC50 on 13/15 targets and much higher internal diversity than 2D Morgan fingerprint search, suggesting the embedding captures binding-relevant geometry rather than just 2D similarity.
6 Because ligands and pockets share the same embedding space, the same system supports ligand-based search for scaffold hopping: given a known active, it retrieves molecules with high 3D shape/pharmacophore similarity but low 2D similarity (lower Tanimoto than Morgan FP), aligning with the goal of finding chemically novel scaffolds that preserve 3D binding features.
7 For generation, they introduce an MCLM (multimodal chemical language model): a Llama2-style autoregressive SMILES decoder conditioned by prepending a learned structural embedding token (from the frozen contrastive encoder) plus a learned dataset token [ds] that controls which chemical space distribution to emulate.
8 The dataset token is the mechanism for “commercial space steering”: trained on a 287M conformer–SMILES corpus labeled by source (PubChem, Enamine Diversity, Mcule, ChEMBL, GEOM-drugs), the model can be prompted with an “Enamine” token at inference to bias outputs toward Enamine-like chemistry; an ablation shows the token increases nearest-neighbor similarity to Enamine REAL and raises the fraction of exact catalog matches.
9 In structure-conditioned de novo design on LIT-PCBA (100 molecules/target), ligand-conditioned generation achieves the highest predicted affinity metrics (affinity probability and predicted pIC50) among evaluated methods, while pocket-conditioned generation (despite the decoder being trained only with ligand embeddings) remains competitive without reference ligands and yields the highest diversity, highlighting practical utility when only target structure is available.
📜Paper: arxiv.org/abs/2604.19562
#ComputationalBiology #Cheminformatics #DrugDiscovery #StructureBasedDrugDesign #GenerativeAI #MachineLearning #ProteinLigand #EquivariantNetworks #ContrastiveLearning #VirtualScreening
3
9
897
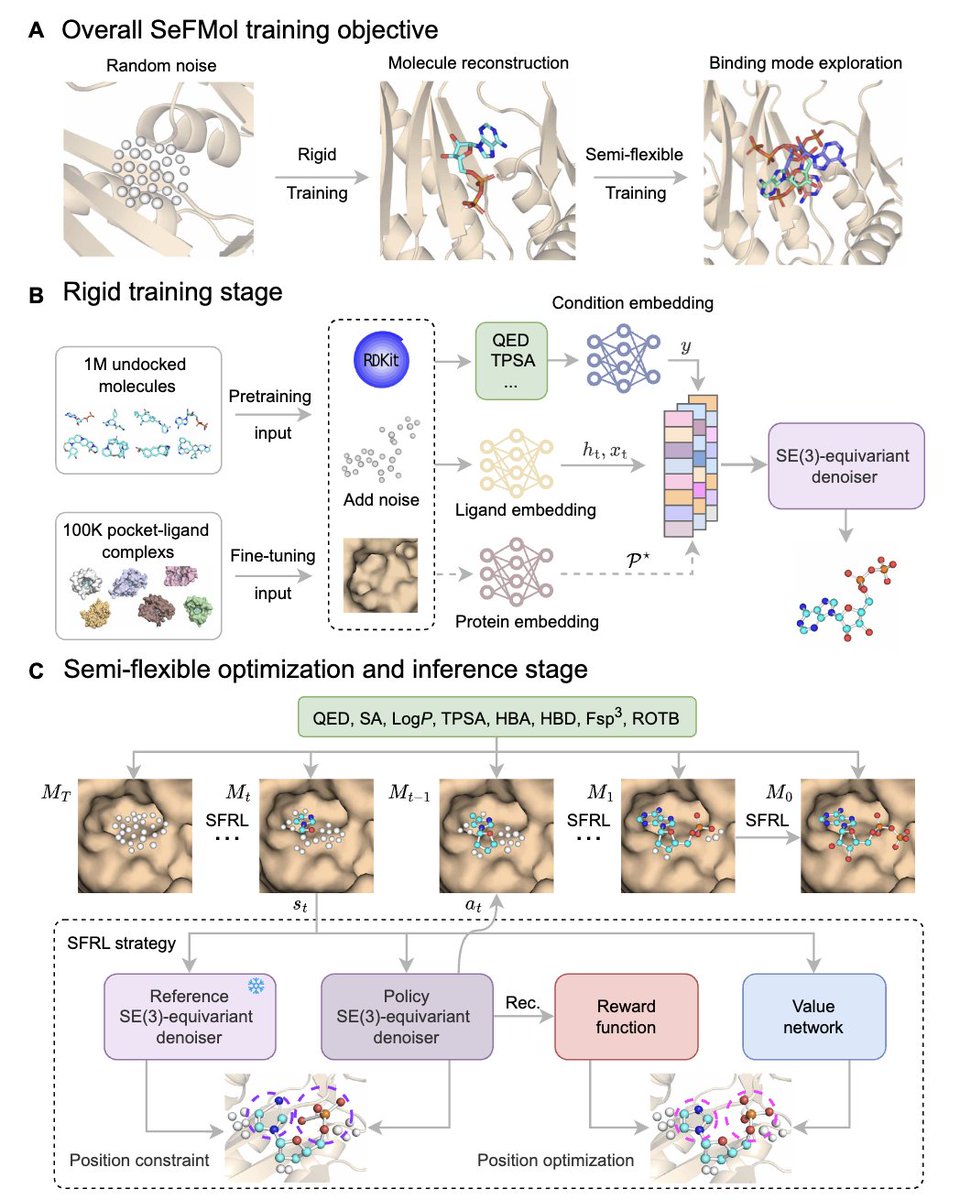
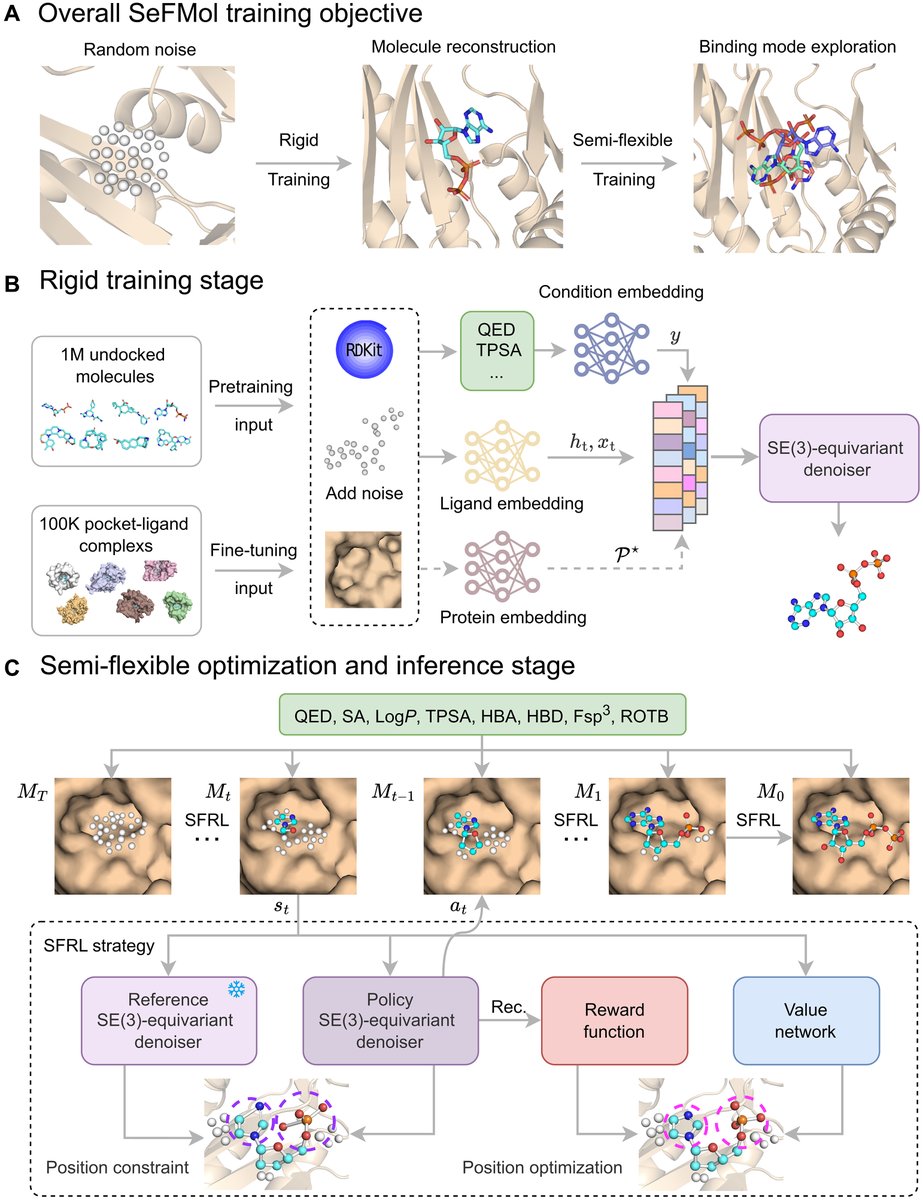
Steering semi-flexible molecular diffusion model for structure-based drug design with reinforcement learning @ScienceAdvances
1. The paper introduces SeFMol, a reinforcement learning (RL)–steered 3D diffusion model that treats diffusion denoising as a Markov decision process, enabling semi-flexible, stepwise conformational adjustment of ligands inside protein pockets rather than assuming rigid ligand conformations.
2. Core idea: a pretrained “rigid” conditional diffusion model is further optimized with a semi-flexible RL stage (SFRL). A frozen reference denoiser anchors the policy denoiser via a KL constraint, helping prevent reward overfitting and catastrophic drift while improving binding-oriented geometry.
3. Property control is built into generation as explicit conditioning signals (computed with RDKit): QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB. These conditions act as dense guidance throughout denoising to counter sparse terminal rewards from docking-based objectives.
4. Training pipeline: (i) pretrain on 1,000,000 target-free Molecule3D molecules with property conditioning to learn general structure–property priors; (ii) fine-tune on 100,000 CrossDocked2020 protein-ligand pairs to become pocket-aware while retaining property bias; (iii) apply SFRL to steer denoising trajectories toward better pocket complementarity.
5. Efficiency contribution: a variable fast sampling strategy reduces diffusion steps from 1000 (training) to 50 (sampling/optimization), yielding ~20x fewer steps while maintaining quality; reported sampling is ~0.81 s per molecule with 98.3% completion, substantially faster than several diffusion baselines.
6. Docking-centric results on 100 test pockets (100 molecules per pocket): SeFMol reports average Vina score −7.23 kcal/mol and improved affinity-related metrics (Vina min and Vina dock). It also reports an SR (success rate under nine joint affinity property constraints) of 11.53%.
7. Geometric/interaction reliability: SeFMol’s generated poses show strong agreement between direct Vina scoring and redocking (reported correlation 0.95), plus competitive RMSD-to-redocked distributions and fewer docking clashes, consistent with the goal of generating chemically plausible 3D conformations directly.
8. Interaction-pattern preservation with optimization: using PLIP interaction typing, SeFMol maintains interaction-type distributions close to reference ligands (reported JSD 0.1401, comparable to the best baseline), suggesting affinity gains do not come from unrealistic interaction artifacts.
9. Generalization case studies: on real-world targets CDK2 (1H00) and ROCK1 (6E9W), SeFMol-generated ligands reproduce canonical interactions seen in known actives while also proposing alternative chemotypes and plausible new interactions; property steering (e.g., setting TPSA or Fsp3 targets) shifts generated distributions around specified values.
📜Paper: doi.org/10.1126/sciadv.ady99…
#ComputationalBiology #StructureBasedDrugDesign #DiffusionModels #ReinforcementLearning #MolecularGeneration #GenerativeAI #DrugDiscovery #GeometricDeepLearning #SE3Equivariance
1
5
15
1,805
Steering semi-flexible molecular diffusion model for structure-based drug design with reinforcement learning
1. The paper presents SeFMol, an RL-steered diffusion framework that treats diffusion denoising as a Markov decision process, enabling “semi-flexible” ligand conformational adjustment inside protein pockets rather than assuming rigid ligands during generation.
2. Core idea: during denoising, a policy denoiser is optimized with reinforcement learning to iteratively refine 3D atom positions (and types) toward better pocket complementarity, while a frozen reference denoiser provides a KL-regularized anchor to prevent the policy from drifting too far and “forgetting” pretrained structure priors.
3. Property-guided generation is built in as conditioning signals to reduce invalid exploration under sparse terminal rewards. SeFMol conditions on eight RDKit properties: QED, SA, LogP, TPSA, HBA, HBD, Fsp3, and ROTB, biasing samples toward drug-like regions while optimizing binding.
4. Training pipeline has two phases: (i) rigid self-supervised diffusion training for molecular reconstruction, then (ii) semi-flexible RL optimization (SFRL). Pretraining uses 1,000,000 target-free molecules from Molecule3D; fine-tuning uses 100,000 protein-ligand pairs from CrossDocked2020 to become pocket-aware.
5. A notable engineering contribution is a variable fast sampling strategy: training uses 1000 diffusion steps, but sampling uses 50 steps (20x fewer). Reported efficiency: ~0.81 s per molecule with 98.3% completion, compared with much slower diffusion baselines (e.g., tens of seconds per molecule for some methods).
6. Docking-focused results on 100 test pockets: SeFMol achieves the best average Vina score (−7.23 kcal/mol) and strong alignment between “direct” Vina score and redocking Vina dock (correlation 0.95), suggesting it often generates chemically plausible poses without relying on redocking to “fix” geometry.
7. Multi-objective quality: SeFMol reports the highest QED among compared methods and competitive SA/Lipinski performance, aiming to avoid the common failure mode where affinity improves but drug-likeness degrades. Property distributions cluster near specified targets (e.g., TPSA control relevant to BBB constraints).
8. Chemical rationality and reliability checks: SeFMol-generated molecules show RMSD distributions closer to reference ligands after redocking and fewer protein-ligand clashes than other methods, consistent with the semi-flexible RL stage learning more stable 3D conformations.
9. Generalization case studies: on real therapeutic targets (CDK2 and ROCK1) and also AlphaFold-predicted structures, SeFMol-generated ligands preserve canonical interaction patterns while also proposing new interaction motifs/chemotypes (e.g., additional halogen bonds or salt bridges in ROCK1 examples), with property control demonstrated by varying TPSA or Fsp3 targets.
📜Paper: doi.org/10.1126/sciadv.ady99…
#StructureBasedDrugDesign #DiffusionModels #ReinforcementLearning #GenerativeModels #ComputationalChemistry #DrugDiscovery #ProteinLigand #SE3Equivariance #MolecularGeneration #AIforScience
4
43
2,073
Apr 12
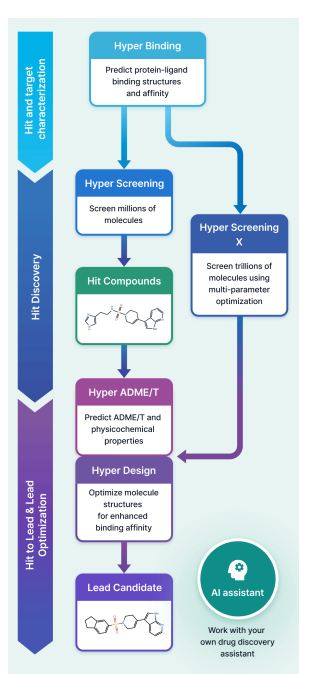
Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
biorxiv.org/content/10.1101/…
Summary:
HyperLab (by HITS) is an AI-driven, web-based drug discovery platform that integrates the full structure-based workflow—from protein–ligand prediction and ultra-large virtual screening (up to trillions of compounds) to molecular design, SAR, and ADME/T prediction—into a single, user-friendly interface. Powered by physics-informed deep learning, it delivers competitive accuracy (approaching AlphaFold3) with significantly faster computation. Experimental validation shows real-world impact, identifying potent compounds (IC50 ~70–600 nM) within 24 hours and enabling rapid optimization. HyperLab lowers the barrier to AI-driven drug discovery by making advanced computational tools accessible to experimental researchers.
#AIDrugDiscovery #HyperLab #StructureBasedDrugDesign #SBDD #DrugDiscovery #ADMET #VirtualScreening #AIBio
1
5
45
2,642
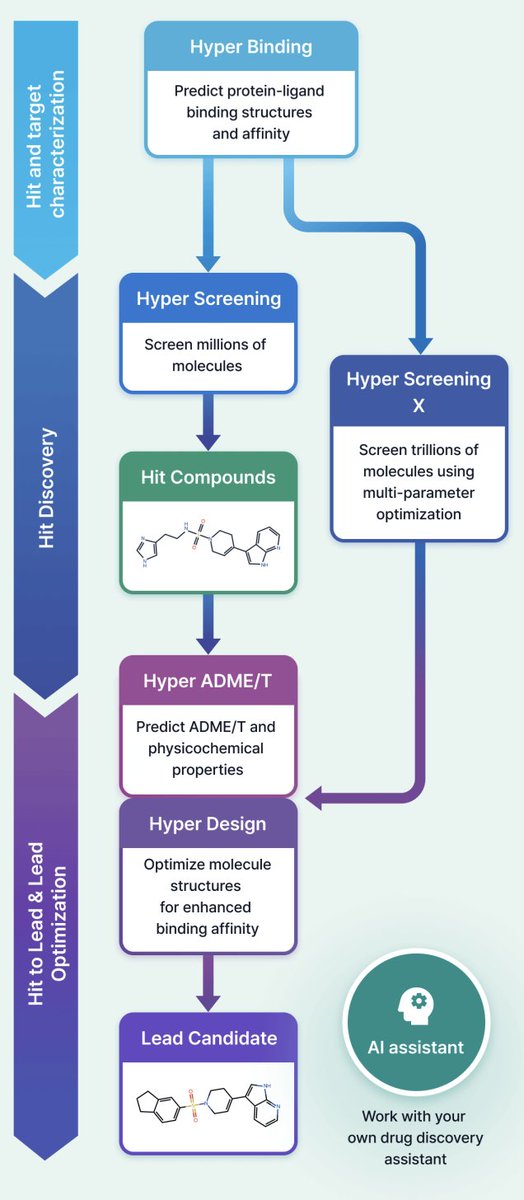
Accelerating Drug Discovery with HyperLab: An Easy-to-Use AI-Driven Platform
1 HyperLab (by HITS) is presented as a web-based, AI-driven SBDD platform aimed at making structure-based workflows usable by experimental drug discovery researchers without requiring AI/CADD expertise, emphasizing integrated UI/UX over fragmented toolchains.
2 The platform compresses early discovery into a single environment spanning: protein–ligand pose affinity prediction (Hyper Binding), covalent complex modeling (Covalent Hyper Binding), virtual screening from 1M to 11T compounds (Hyper Screening / Hyper Screening X), structure-based molecular optimization (Hyper Design), SAR analysis, and 19-endpoint ADME/T prediction (Hyper ADME/T), with an embedded AI assistant for workflow automation.
3 Hyper Binding’s key technical angle is physics-informed deep learning for protein–ligand interactions, supporting multiple protein inputs (PDB ID, uploaded PDB, AlphaFold structures via UniProt) and an end-to-end co-folding mode that predicts complex structures directly from protein sequence plus ligand, reducing dependence on curated receptor structures.
4 On PoseBuster v2 (PB-valid) pose prediction, Hyper Binding reports 77% accuracy when given binding-site information, compared with 58% for Vina and 13% for DiffDock; it approaches AlphaFold3 (84%) and is comparable to Boltz2 (78). The paper also highlights throughput: ~3 minutes per complex (via cloud) vs ~15 minutes for AlphaFold3 on an RTX 3060.
5 For binding affinity prediction on two FEP-style benchmarks (focused on subtle potency differences among close analogs), Hyper Binding reports Pearson r = 0.70 and 0.53, outperforming evaluated deep learning scorers (Luminet, GenScore) and physics-based docking (Glide SP, Vina) on both datasets.
6 Covalent drug discovery is treated as a first-class workflow: covalent pose prediction is benchmarked on a curated covalent set (from PDBBind/PDB). Covalent Hyper Binding (cofolding) reports 88.7% pose accuracy vs 48.4% (COV SMINA) and 46.8% (GNINA); the docking mode reports 61.3%. Screening enrichment (EF@10%) is reported as 6.56 (Mpro) and 9.97 (KRAS), exceeding baselines under the described setup.
7 Hyper Screening targets rapid hit finding by running Hyper Binding across curated libraries and returning top-ranked candidates (top 500). Built-in libraries include: Diverse (1,000,000), Fragment (500,000; rule-of-three-like), Kinase-focused (65,000), Natural product-like fragments (4,200), and FDA-approved (1,100), plus support for user-registered libraries.
8 Hyper Screening X expands to an 11-trillion-molecule virtual space using generative exploration with GFlowNet-based models, optimizing binding score plus properties (e.g., MW, TPSA, LogP). The workflow is described as: set target property constraints, train (~48h), then generate molecules (e.g., 100 molecules in ~30 min), with synthetic route output and optional synthesis request via a partner service.
9 Hyper Design provides structure-based optimization starting from a scaffold or an X-ray-bound ligand, enabling user-specified modification sites and fragment growth/replacement with synthesizability constraints; outputs include 3D structures and iterative “design trees.” The paper positions use cases as fragment-to-lead growth and generating patent-distinct analogs while preserving key interactions.
10 The internal validation study emphasizes “no post-analysis/visual inspection” selection: a 24-hour Hyper Screening run led to 52 compounds tested, yielding 5 hits with IC50 70–600 nM (~9% hit rate). Hyper Design then produced derivatives; 5 were synthesized and 3 showed >75% inhibition at 1 µM with IC50 200–400 nM, including one compound comparable or better than a reference and with supporting pathway assay readouts.
📜Paper: biorxiv.org/content/10.1101/…
#DrugDiscovery #ComputationalBiology #Cheminformatics #StructureBasedDrugDesign #VirtualScreening #CovalentInhibitors #ADMET #GenerativeAI #ProteinLigandDocking #BioRxiv
3
16
1,462
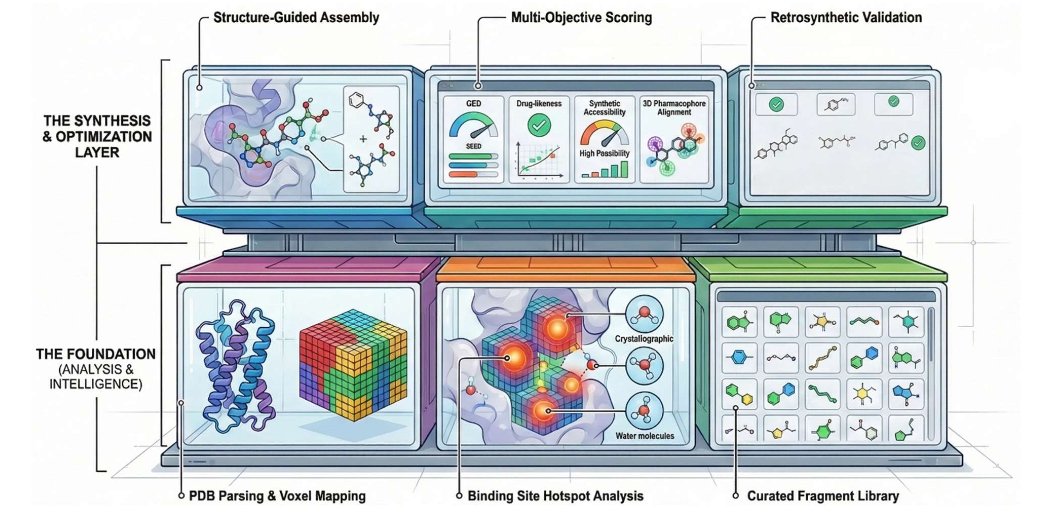
LigandForge: A Web Server for Structure-Guided De Novo Drug Design
1 LigandForge is presented as an end-to-end, browser-based workflow for structure-guided de novo ligand design that explicitly couples pocket physics (3D voxel fields hotspots water thermodynamics) with synthesis awareness (retrosynthetic feasibility), aiming to reduce both licensing and programming barriers.
2 The core technical idea is a voxel-based property grid around the binding site: electrostatics, hydrophobicity, steric accessibility/excluded volume, openness/SASA-derived cavity boundaries, plus geometric descriptors (curvature/shape index). These fields provide directional gradients used to guide fragment placement and growth in 3D.
3 A notable feature is explicit handling of crystallographic waters: each water site is assigned replaceability and entropy/energy-related contributions, and high-energy waters are flagged as displacement targets to guide ligand functionalization with thermodynamic rationale.
4 Molecules are built via chemistry-aware fragment assembly from curated libraries organized by functional role (core scaffolds, linkers, substituents, bioisosteres). An attachment-point manager checks local hybridization (sp/sp2/sp3), aromaticity, and valence, while fragments are oriented using local field gradients to match pocket interaction demands.
5 Drug-likeness constraints are enforced during growth (not only after generation): MW 150–700 Da, LogP −4 to 7, rotatable bond limits, and heavy atom count constraints (10–50). This keeps candidates within practical physicochemical ranges throughout the assembly trajectory.
6 Multi-objective optimization combines pharmacophore/hotspot alignment, QED-based drug-likeness, synthetic accessibility, and novelty/diversity into a weighted composite score with target-class presets (e.g., kinase vs GPCR) that adjust weights for different binding-site archetypes.
7 Optimization supports reinforcement learning, genetic algorithms, and hybrid heuristics; chemical space coverage is maintained using fingerprint-based diversity control with DBSCAN clustering to reduce mode collapse and encourage scaffold variety.
8 Synthesis awareness is integrated as a hard filter/penalty: a retrosynthetic analyzer decomposes top candidates into routes and assigns difficulty, step/yield proxies, and feasibility. The final reward is scaled by route feasibility and penalized by difficulty, pushing optimization away from impractical structures.
9 Prospective evaluation used Boltz-2 co-folding/affinity prediction on three targets (D2R, EGFR, STAT5b-NTD). Across 10 runs per target (100 candidates/target), candidates reached micromolar predicted affinities, with submicromolar predictions for D2R; predicted poses recapitulated key motif interactions (D2R D3.32 salt bridge; EGFR hinge H-bonding in the ATP site).
10 Novelty was assessed against known actives (ChEMBL for D2R/EGFR; limited literature set for STAT5b-NTD): every generated molecule reportedly had Tanimoto similarity < 0.3 to the closest known active for its target, suggesting exploration of distinct chemotypes while retaining predicted binding competence.
💻Code: github.com/HTS-Oracle/Ligand…
📜Paper: biorxiv.org/content/10.64898…
#DeNovoDesign #FragmentBasedDrugDesign #StructureBasedDrugDesign #Cheminformatics #RDKit #ReinforcementLearning #GeneticAlgorithms #Retrosynthesis #WebServer #DrugDiscovery
1
5
39
3,117
BioLM-Score: Language-Prior Conditioned Probabilistic Geometric Potentials for Protein-Ligand Scoring
1. The authors introduce BioLM-Score, a novel protein-ligand scoring framework that bridges geometric deep learning with biomolecular representation learning, achieving state-of-the-art performance across scoring, ranking, docking, and virtual screening tasks on CASF-2016 and DEKOIS 2.0 benchmarks.
2. The key innovation lies in integrating pre-trained biomolecular language models (ESM-C for proteins and Chemformer for ligands) with structure-aware graph encoders, enabling the model to condition local geometric interactions on global evolutionary and chemical semantics.
3. Unlike conventional approaches that rely solely on 3D graphs, this dual-branch architecture enriches geometric representations with sequence-based priors, addressing the critical limitation of overlooking global contexts in existing geometric likelihood methods.
4. The model employs a mixture density network to predict multimodal interatomic distance distributions, formulating the final score as an aggregated log-likelihood that captures probabilistic geometric potential while maintaining physical interpretability.
5. Three training variants are proposed: a geometry-only baseline, joint training with affinity supervision, and a two-stage fine-tuning protocol that effectively balances geometric consistency with binding affinity prediction without optimization conflicts.
6. Ablation studies reveal that protein language model features play a pivotal role in docking and screening performance, while ligand language priors provide complementary benefits for rebalancing optimization objectives across multiple tasks.
7. The scoring function demonstrates practical utility as a differentiable optimization objective within the BSDock framework, guiding conformational search to achieve a 71.58% docking success rate on CASF-2016 when combined with local refinement.
📜Paper: arxiv.org/abs/2602.18476
#BioLMScore #ProteinLigandScoring #StructureBasedDrugDesign #GeometricDeepLearning #ProteinLanguageModels #VirtualScreening #MolecularDocking #DrugDiscovery #ComputationalBiology

1
19
1,839
EnzyPGM: Pocket-conditioned Generative Model for Substrate-specific Enzyme Design
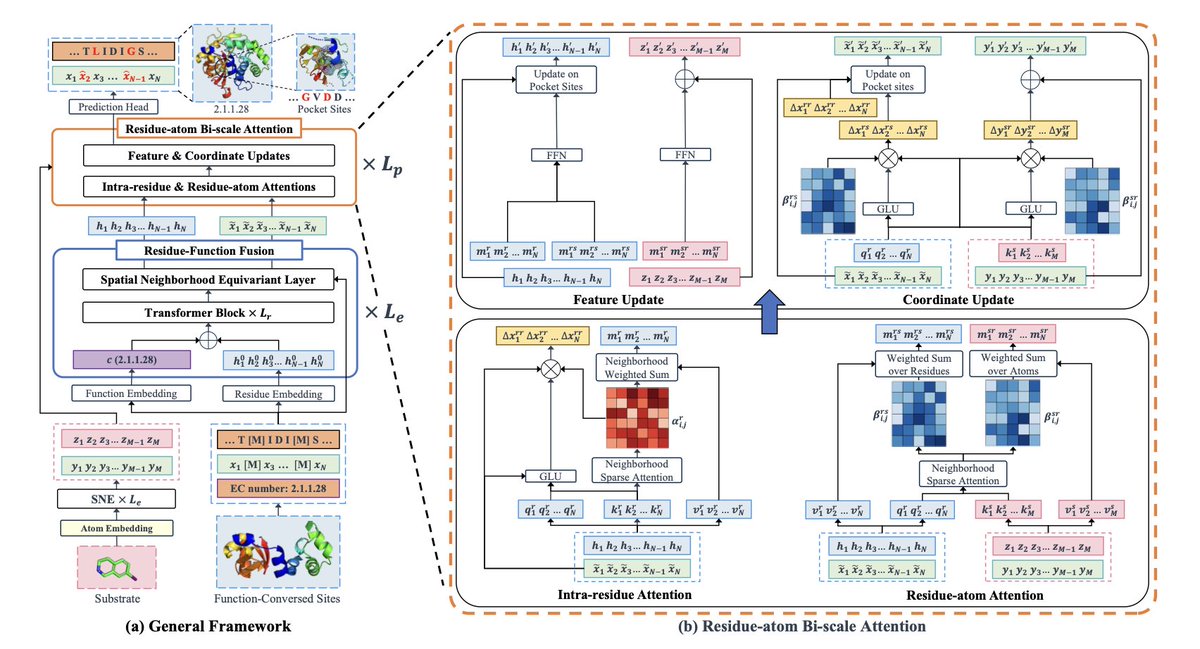
1 EnzyPGM is the first unified model that jointly generates the full enzyme sequence and its 3-D substrate-binding pocket, explicitly conditioned on the substrate molecule and the enzyme’s EC number.
2 A Residue-atom Bi-scale Attention (RBA) module performs simultaneous intra-residue and residue–substrate attention, letting the network learn fine-grained pocket-substrate geometries and avoid steric clashes.
3 Residue Function Fusion (RFF) injects EC-based functional priors and evolutionarily conserved residues into the embedding, guiding the generator toward catalytically competent motifs while preserving SE(3) equivariance.
4 Training on the newly curated EnzyPock data set (83 k enzyme–substrate pairs, 1 036 EC-4 families) yields state-of-the-art AAR (0.77) and a Vina score 0.47 kcal mol⁻¹ better than the previous best method, EnzyGen.
5 Ablation shows that removing either attention stream drops pLDDT by 1–2 points, confirming that explicit pocket-substrate interaction modeling is the key driver of improved binding affinity and foldability.
6 Case visualizations reveal that EnzyPGM pockets contain more hydrogen bonds and π-stacking contacts than baseline-generated pockets, explaining the lower Vina scores and higher substrate specificity.
7 t-SNE of latent representations clusters enzymes by EC family, indicating the model learns function-aware embeddings that generalize to unseen EC numbers and novel substrates without retraining.
📜Paper: arxiv.org/abs/2601.19205
#enzymeDesign #proteinEngineering #generativeAI #structureBasedDrugDesign #bioinformatics
3
21
1,524
Beyond Affinity: A Benchmark of 1D, 2D, and 3D Methods Reveals Critical Trade-offs in Structure-Based Drug Design
1. A new benchmark study evaluates 15 different models across 1D, 2D, and 3D methods in structure-based drug design, highlighting critical trade-offs between binding affinity, chemical validity, and pose quality. This comprehensive analysis fills a gap in cross-algorithm comparisons and offers insights for future model development.
2. The study finds that 3D models excel in binding affinity predictions but show inconsistencies in chemical validity and pose quality. In contrast, 1D models demonstrate reliable performance in standard molecular metrics but rarely achieve optimal binding affinities. 2D models offer a balanced performance, maintaining high chemical validity while achieving moderate binding scores.
3. A key innovation is the use of detailed pose quality assessments, including PoseBuster and PoseCheck evaluations, alongside geometric consistency checks. This allows for a focused diagnosis of the "affinity-validity trade-off," providing a more nuanced understanding of model strengths and weaknesses.
4. The research highlights that 1D/2D ligand-centric methods can be effectively used in structure-based drug design by treating the docking function as a black-box oracle. This approach is often neglected but shows promise for efficient molecule generation.
5. The study emphasizes the need for future models to better balance docking score optimization with other crucial molecular properties and structural validity. It identifies key areas for improvement and suggests that hybrid models combining the strengths of different approaches could be a promising direction.
📜Paper: arxiv.org/abs/2601.14283v1
#MachineLearning #DrugDesign #StructureBasedDrugDesign #Benchmarking #AIinMedicine
4
16
1,468

5 Dec 2025
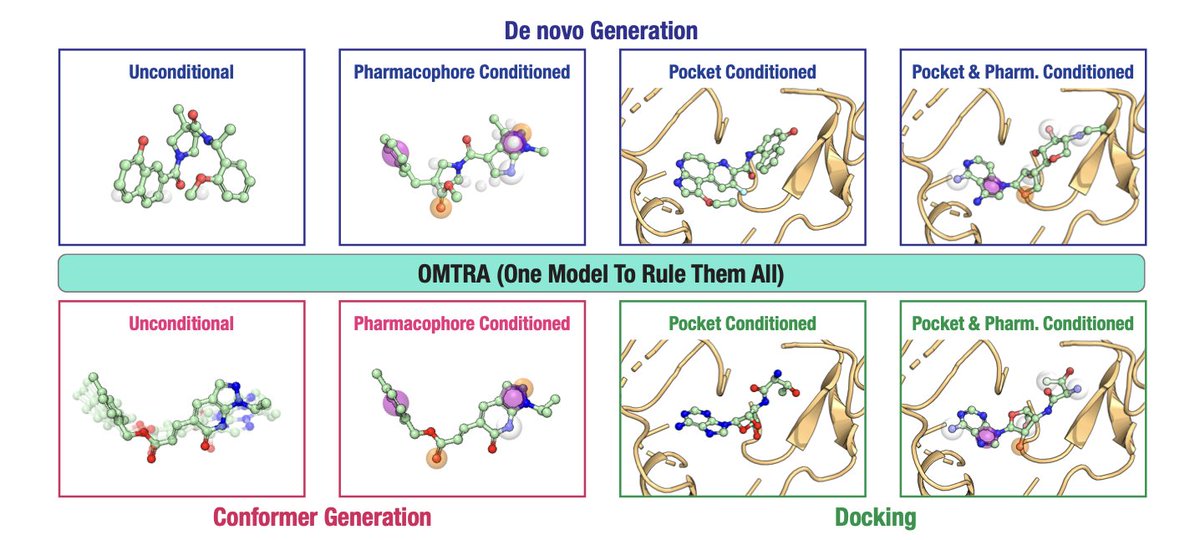
OMTRA: A Multi-Task Generative Model for Structure-Based Drug Design
1. OMTRA introduces a novel multi-task generative model that unifies various tasks in structure-based drug design (SBDD) under one framework. This includes de novo ligand design, docking, and conformer generation, showcasing versatility in handling multiple SBDD-related tasks.
2. The model leverages flow matching, a powerful technique for interpolating between probability distributions, to generate molecular structures. This approach enables the simultaneous modeling of continuous and discrete molecular properties, such as atom positions and types.
3. A key innovation is the ability to condition the model on protein pockets and pharmacophores, allowing for guided design and docking. This feature enhances the accuracy of ligand generation by incorporating prior knowledge of protein-ligand interactions.
4. OMTRA achieves state-of-the-art performance in pocket-conditioned de novo design and docking, outperforming existing models in terms of both chemical plausibility and interaction recovery with proteins.
5. The authors curate a large-scale dataset of 500 million 3D molecular conformers, significantly expanding the chemical diversity available for training. This dataset complements existing protein-ligand data and supports multi-task learning across diverse molecular modalities.
6. Despite the potential benefits of multi-task training, the study finds that its effects are modest and sometimes inconsistent across different tasks. This highlights the ongoing challenge of effectively leveraging transfer learning in molecular generative models.
7. Future work will explore extending OMTRA to generate protein structures, which could support tasks involving flexible or unknown protein conformations. This extension could make OMTRA a more comprehensive tool for realistic drug design scenarios.
💻Code: github.com/gnina/OMTRA
📜Paper: arxiv.org/abs/2512.05080
#DrugDesign #GenerativeModeling #FlowMatching #MultiTaskLearning #StructureBasedDrugDesign #MolecularGeneration
1
4
28
2,195